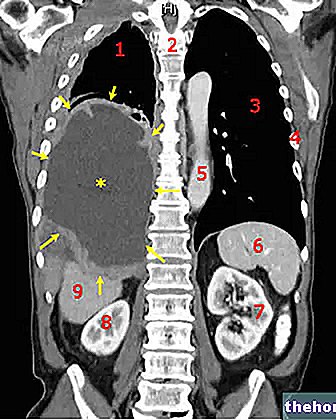
Účinné látky: Dabigatran (dabigatran etexilát)
Pradaxa 150 mg tvrdé tobolky
Příbalové letáky Pradaxa jsou k dispozici pro velikosti balení:- Pradaxa 75 mg tvrdé tobolky
- Pradaxa 110 mg tvrdé tobolky
- Pradaxa 150 mg tvrdé tobolky
Proč se přípravek Pradaxa používá? K čemu to je?
Pradaxa je léčivý přípravek, který obsahuje léčivou látku dabigatran -etexilát. Blokuje působení látky v těle, která se podílí na tvorbě krevních sraženin.
Pradaxa je léčivý přípravek používaný ke snížení rizika zablokování krevních cév v mozku nebo zbytku těla tvorbou krevních sraženin u dospělých pacientů se změněným srdečním tepem (fibrilace síní) as dalšími rizikovými faktory. Pradaxa je lék, který ředí krev a snižuje riziko vzniku krevních sraženin.
Pradaxa je lék používaný k léčbě krevních sraženin v žilách nohou a plic a k prevenci návratu krevních sraženin v žilách nohou a plic.
Kontraindikace Kdy by Pradaxa neměla být použita
Neužívejte přípravek Pradaxa
- jestliže jste alergický (á) na dabigatran -etexilát nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže máte závažně sníženou funkci ledvin
- pokud máte trvalé krvácení.
- jestliže máte poranění orgánu, které zvyšuje riziko závažného krvácení.
- jestliže máte zvýšenou tendenci ke krvácení.To může být vrozené, z neznámé příčiny nebo z důvodu jiných léků.
- jestliže máte výrazně sníženou funkci jater nebo onemocnění jater, které může nějakým způsobem způsobit smrt.
- pokud užíváte ketokonazol nebo itrakonazol ústy, léky k léčbě plísňových infekcí.
- jestliže užíváte cyklosporin, lék k prevenci epizod odmítnutí po transplantaci orgánu.
- pokud užíváte dronedaron, lék používaný k prevenci návratu problému s nepravidelným srdečním tepem.
- pokud užíváte léky k prevenci tvorby krevních sraženin (např. warfarin, rivaroxaban, apixaban nebo heparin), kromě případů, kdy přecházíte z jedné antikoagulační léčby na jinou nebo když jste umístili arteriální žilní katétr a vezměte jím heparin, aby byl otevřený.
- pokud vám byla implantována umělá srdeční chlopně.
Opatření pro použití Co potřebujete vědět před užitím přípravku Pradaxa
Před užitím přípravku Pradaxa se poraďte se svým lékařem. Také můžete během léčby přípravkem Pradaxa navštívit svého lékaře, pokud se u vás objeví příznaky nebo pokud potřebujete operaci. Informujte svého lékaře, pokud máte nebo jste trpěl (a) jakýmkoli zdravotním stavem nebo nemocí, zejména některým z těch, které jsou uvedeny v následujícím seznamu:
- pokud trpíte onemocněním jater spojeným s abnormálními krevními testy, používání přípravku Pradaxa se nedoporučuje.
- pokud máte zvýšené riziko krvácení, které by mohlo být v následujících situacích:
- jestliže jste nedávno prodělal krvácení.
- pokud jste v předchozím měsíci podstoupili biopsii (chirurgické odstranění tkáně).
- pokud jste utrpěli vážná zranění (např. zlomeninu kosti, poranění hlavy nebo jakékoli zranění, které si vyžádalo chirurgický zákrok).
- jestliže trpíte zánětem jícnu nebo žaludku.
- jestliže máte problémy s refluxem žaludeční šťávy do jícnu.
- pokud jste užívali léky, které mohou zvyšovat riziko krvácení, jako je aspirin (kyselina acetylsalicylová), klopidogrel, tikagrelor.
- pokud užíváte protizánětlivé léky, jako je diklofenak, ibuprofen, piroxikam.
- jestliže máte srdeční infekci (bakteriální endokarditidu).
- pokud víte, že máte poškozenou funkci ledvin nebo trpíte dehydratací (příznaky zahrnují pocit žízně a močení ve sníženém množství tmavé (koncentrované) moči).
- pokud je vám více než 75 let.
- pokud váží 50 kg nebo méně.
- jestliže jste prodělal (a) srdeční infarkt nebo vám byly diagnostikovány stavy, které zvyšují riziko vzniku srdečního záchvatu.
- pokud podstupujete plánovanou operaci. Přípravek Pradaxa bude nutné dočasně vysadit kvůli zvýšenému riziku krvácení během operace a krátce po ní. Pokud je to možné, Pradaxa by měla být zastavena nejméně 24 hodin před operací. U pacientů se zvýšeným rizikem krvácení se lékař může rozhodnout ukončit léčbu dříve.
- pokud podstupujete neplánovanou operaci. Pokud je to možné, chirurgický zákrok by měl být odložen na 12 hodin po poslední dávce přípravku Pradaxa. Pokud operaci nelze odložit, může dojít ke zvýšenému riziku krvácení.Váš lékař posoudí riziko krvácení a naléhavost operace.
- pokud máte v zádech zavedenou hadičku (katétr): trubici lze vložit do zad, např. k podávání anestetik nebo léků tlumících bolest během nebo po chirurgickém zákroku.Pokud dostanete přípravek Pradaxa po odstranění katétru, bude vás lékař pravidelně kontrolovat.
- pokud během léčby upadnete nebo se zraníte, zvláště pokud dostanete ránu do hlavy, okamžitě zavolejte svého lékaře. Váš lékař může považovat za nutné, aby vás viděl, protože vám může hrozit vysoké riziko krvácení.
Děti a dospívající
Přípravek Pradaxa by neměly používat děti a mladiství do 18 let.
Interakce Které léky nebo potraviny mohou změnit účinek přípravku Pradaxa
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval (a) nebo které možná budete užívat. Např:
- Léky snižující srážlivost krve (např. Warfarin, fenprokumon, heparin, klopidogrel, prasugrel, tikagrelor, rivaroxaban)
- Protizánětlivé a bolest tlumící léky (např. Aspirin)
- Třezalka tečkovaná, bylinný přípravek k léčbě deprese
- Antidepresiva nazývaná selektivní inhibitory zpětného vychytávání serotoninu nebo selektivní inhibitory zpětného vychytávání serotoninu a norepinefrinu
- Rifampicin nebo klarithromycin, dvě antibiotika
- Léky k léčbě změněného srdečního tepu (např. Amiodaron, dronedaron, chinidin, verapamil). Pokud užíváte léky obsahující verapamil, měli byste užívat sníženou dávku 220 mg Pradaxy v jedné kapsli 110 mg dvakrát denně, protože se může zvýšit riziko krvácení.Pradaxa a léky obsahující verapamil by měly být užívány společně.
- Léky k léčbě plísňových infekcí (např. Ketokonazol, itrakonazol, posakonazol), pokud nejsou aplikovány pouze na kůži
- Léky k prevenci přímých epizod po transplantaci orgánů (např. Takrolimus, cyklosporin)
- Virové léky na AIDS (např. Ritonavir)
- Léky k léčbě epilepsie (např. Karbamazepin, fenytoin)
Varování Je důležité vědět, že:
Těhotenství a kojení
Účinky přípravku Pradaxa na těhotenství a plod nejsou známy. Pokud jste těhotná, nesmíte přípravek Pradaxa užívat, pokud vám lékař neřekne, že je to bezpečné. Pokud jste žena ve fertilním věku, musíte během léčby přípravkem Pradaxa zabránit otěhotnění.
Během léčby přípravkem Pradaxa byste neměla kojit.
Řízení dopravních prostředků a obsluha strojů
Pradaxa nemá žádné známé účinky na schopnost řídit nebo obsluhovat stroje.
Pradaxa obsahuje oranžovou žluť (E110)
Tento léčivý přípravek obsahuje barvivo zvané oranžová žlutá (E110), které může způsobit alergické reakce
Dávka, způsob a doba podání Jak se přípravek Pradaxa používá: Dávkování
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud máte pochybnosti, poraďte se se svým lékařem.
Vezměte Pradaxu podle doporučení pro následující podmínky:
Prevence tvorby krevních sraženin po operaci náhrady kolene nebo kyčle
Doporučená dávka je 300 mg jednou denně (užívá se jako 2 tobolky po 150 mg).
Pokud je vaše funkce ledvin snížena o více než polovinu nebo je vám 75 nebo více let, doporučená dávka je 220 mg užitá jako jedna 110mg tobolka dvakrát denně.
Pokud užíváte léky obsahující verapamil, měli byste užívat sníženou dávku 220 mg Pradaxy v jedné kapsli 110 mg dvakrát denně, protože se může zvýšit riziko krvácení.
Pokud máte potenciálně zvýšené riziko krvácení, může se váš lékař rozhodnout předepsat dávku 220 mg přípravku Pradaxa užívanou jako jednu 110mg tobolku dvakrát denně.
Přípravek Pradaxa lze užívat s jídlem nebo bez jídla. Tobolka by měla být spolknuta celá se sklenicí vody, aby se zajistilo uvolňování žaludku.Nerozlomte, nežvýkejte ani nevyjímejte granule z tobolky, protože to může zvýšit riziko krvácení.
Při používání přípravku Pradaxa v blistru dodržujte následující pokyny
- vyjměte tobolky z blistru zvednutím hliníkové fólie na zadní straně.
- kapsle netlačte přes blistr.
- hliníková fólie z blistru by měla být zvednuta pouze tehdy, když má být vyjmuta tobolka.
Při používání přípravku Pradaxa zabaleného v lahvičce dodržujte následující pokyny
- láhev se otevírá stisknutím a otočením víčka.
Změna v antikoagulační léčbě
- Přechod z léčby přípravkem Pradaxa na léčbu antikoagulancii podávanými injekčně: Nezačínejte léčbu injekčními antikoagulačními léky (např. Heparinem), dokud od posledního podání přípravku Pradaxa neuplyne 12 hodin.
- Přechod z léčby antikoagulancii podávanými injekcí na léčbu přípravkem Pradaxa: Začněte užívat přípravek Pradaxa 0–2 hodiny před časem další injekce.
- Přechod z léčby přípravkem Pradaxa na léčbu léky na ředění krve obsahujícími antagonisty vitaminu K (např. Fenprokumon): lékař by vás měl požádat o vyšetření krve a sdělit vám, kdy zahájit léčbu antagonisty vitaminu K.
- Přechod z léčby léky na ředění krve obsahujícími antagonisty vitaminu K (např. Fenprokumon) na léčbu přípravkem Pradaxa: Přestaňte užívat lék obsahující antagonistu vitaminu K. Váš lékař by vás měl požádat o vyšetření krve a sdělit vám, kdy zahájíte léčbu přípravkem Pradaxa.
Předávkování Co dělat, pokud jste užili příliš mnoho přípravku Pradaxa
Jestliže jste užil (a) více přípravku Pradaxa, než jste měl (a)
Pokud užijete více přípravku Pradaxa, než je doporučeno, můžete mít zvýšené riziko krvácení. Váš lékař může provést krevní test, aby vyhodnotil riziko krvácení.
Okamžitě informujte svého lékaře, pokud užijete více přípravku Pradaxa, než je předepsáno. Pokud dojde k epizodě krvácení, může být nutná operace nebo léčba transfuzí krve.
Jestliže jste zapomněl (a) užít přípravek Pradaxa
Vynechanou dávku je možné užít až 6 hodin před další dávkou. Pokud je před další dávkou méně než 6 hodin, vynechanou dávku je třeba přeskočit. Nezdvojnásobujte následující dávku, abyste nahradil (a) vynechanou dávku
Jestliže jste přestal (a) užívat přípravek Pradaxa
Užívejte přípravek Pradaxa přesně podle předpisu. Nepřestávejte užívat přípravek Pradaxa bez předchozí kontroly u svého lékaře.Ukončení léčby přípravkem Pradaxa může zvýšit riziko vzniku krevní sraženiny u pacientů léčených po operaci náhrady kyčle nebo kolena.
Máte -li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Nežádoucí účinky Jaké jsou vedlejší účinky přípravku Pradaxa
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pradaxa působí na systém srážení krve, takže většina vedlejších účinků souvisí s příznaky, jako je hematom nebo krvácení.
Mohou nastat závažné nebo závažné krvácivé příhody, jedná se o nejzávažnější nežádoucí účinky, které bez ohledu na místo mohou být invalidizující, život ohrožující nebo dokonce vést k úmrtí. V některých případech nemusí být toto krvácení evidentní.
Pokud se u vás objeví jakékoli krvácení, které spontánně nevymizí, nebo pokud se u vás objeví příznaky nadměrného krvácení (výjimečná slabost, pocit únavy, bledá kůže, závratě, bolesti hlavy nebo nevysvětlitelný otok), okamžitě navštivte svého lékaře.
Váš lékař může rozhodnout o pečlivé kontrole nebo o změně léčby.
Okamžitě informujte svého lékaře, pokud se u vás objeví závažná alergická reakce, která způsobuje potíže s dýcháním nebo závratě.
Nežádoucí účinky jsou uvedeny níže, seskupené podle toho, jak často se vyskytují.
Prevence zablokování krevních cév v mozku nebo zbytku těla v důsledku tvorby krevních sraženin, které se vyvinuly v důsledku změněného srdečního tepu
Časté (mohou postihnout až 1 z 10 lidí):
- Krvácení, které se může objevit z nosu, žaludku nebo střev, penisu / pochvy nebo močových cest (včetně krve v moči, která je růžová nebo červená) nebo pod kůží
- Snížení počtu červených krvinek v krvi
- Bolest břicha nebo žaludku
- Špatné trávení
- Průjem se špatně tvarovanou nebo tekutou stolicí
- Necítím se dobře
Méně časté (mohou postihnout až 1 ze 100 lidí):
- Krvácející
- Krvácení, které může nastat z hemoroidů, do konečníku nebo do mozku
- Tvorba hematomu
- Vykašlávání krve nebo krvavě zbarveného sputa
- Snížení počtu krevních destiček v krvi
- Snížení množství hemoglobinu v krvi (látka nacházející se v červených krvinkách)
- Alergická reakce
- Náhlá změna pokožky, která mění její barvu a vzhled
- Svědění
- Gastrointestinální vřed (včetně vředu jícnu)
- Zánět jícnu a žaludku
- Reflux žaludečních šťáv do jícnu
- Zvracel
- Obtížné polykání
- Abnormální výsledky testů jaterních funkcí
Vzácné (mohou postihnout až 1 z 1000 lidí):
- Krvácení, které může nastat v kloubu, z chirurgického řezu, z rány, z místa vpichu nebo z místa zavedení katétru do žíly - Závažná alergická reakce způsobující potíže s dýcháním nebo závratě - Těžká alergická reakce způsobující otok obličeje nebo kožní vyrážka patrná z tmavě červené, oteklé, svědivé hrudky způsobené alergickou reakcí - snížení podílu červených krvinek - zvýšení jaterních enzymů - zežloutnutí kůže nebo očního bělma při problémech s játry nebo krví
Není známo (frekvenci nelze z dostupných údajů určit):
- Obtížné dýchání nebo sípání
Léčba krevních sraženin v žilách nohou a plic včetně prevence opětovné tvorby krevních sraženin v žilách nohou a / nebo plic
Časté (mohou postihnout až 1 z 10 lidí):
- Krvácení, které se může objevit z nosu, žaludku nebo střev, konečníku, penisu / pochvy nebo močových cest (včetně krve v moči, která je růžová nebo červená) nebo pod kůží
- Špatné trávení
Méně časté (mohou postihnout až 1 ze 100 lidí):
- Krvácející
- Krvácení, které může nastat v kloubu z rány
- Krvácení, ke kterému může dojít z hemoroidů
- Snížení počtu červených krvinek v krvi
- Tvorba hematomu
- Vykašlávání krve nebo sputa zbarvené krví
- Alergická reakce
- Náhlá změna pokožky, která mění její barvu a vzhled
- Svědění
- Gastrointestinální vřed
- Zánět jícnu a žaludku
- Reflux žaludečních šťáv do jícnu
- Necítím se dobře
- Zvracel
- Bolest břicha nebo žaludku
- Průjem se špatně tvarovanou nebo tekutou stolicí
- Obtížné polykání
- Abnormální výsledky testů jaterních funkcí
- Zvýšené jaterní enzymy
Vzácné (mohou postihnout až 1 z 1000 lidí):
- Krvácení, které může nastat, buď z „chirurgického řezu nebo z místa vpichu nebo z místa zavedení katétru do žíly nebo z mozku
- Snížení počtu krevních destiček v krvi
- Těžká alergická reakce způsobující potíže s dýcháním nebo závratě
- Těžká alergická reakce, která způsobuje otok obličeje nebo hrdla
- Znatelná kožní vyrážka s tmavě červenými, oteklými, svědivými hrudkami způsobenými alergickou reakcí
- Obtížné polykání
- Snížení počtu červených krvinek v krvi
Není známo (frekvenci nelze z dostupných údajů určit):
- Obtížné dýchání nebo sípání
- Snížení množství hemoglobinu přítomného v krvi (látka obsažená v červených krvinkách)
- Snížení počtu červených krvinek v krvi
- Žloutnutí kůže nebo očního bělma, způsobené problémy s játry nebo krví
Hlášení nežádoucích účinků
Pokud se u vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. To se týká i nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení.
V klinické studii byla frekvence srdečních záchvatů vyšší u přípravku Pradaxa než u warfarinu. Celkový výskyt byl nízký.
Expirace a retence
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce, blistru nebo lahvičce za EXP. Datum exspirace se vztahuje k poslednímu dni uvedeného měsíce.
Blistr: Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Láhev: Po otevření by měl být přípravek spotřebován do 4 měsíců. Láhev uchovávejte těsně uzavřenou. Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Nevyhazujte žádné léky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Pomůže to chránit životní prostředí.
Složení a léková forma
Co přípravek Pradaxa obsahuje
- Léčivou látkou je dabigatran, který se podává ve formě dabigatranetexilátu jako mesylátu dabigatranetexilátu v dávce 150 mg.
- Dalšími složkami jsou kyselina vinná, arabská guma, hypromelóza, dimethikon 350, mastek a hydroxypropylcelulóza.
- Obal tobolky obsahuje karagenan, chlorid draselný, oxid titaničitý, indigokarmín, oranžovou žluť (E110), hypromelózu a čištěnou vodu.
- Černý tiskový inkoust obsahuje šelak, N-butylalkohol, isopropylalkohol, průmyslově denaturovaný ethanol, černý oxid železitý, čištěnou vodu a propylenglykol.
Popis toho, jak Pradaxa vypadá, a obsah balení
Pradaxa je tvrdá tobolka.
Pradaxa 150mg tvrdé tobolky mají neprůhledné světle modré víčko a neprůhledné krémové tělo. Logo Boehringer Ingelheim je vytištěno na víčku a kód „R150“ na těle kapsle.
Pradaxa 150mg tvrdé tobolky jsou k dispozici v baleních obsahujících 10x1, 30x1, 60x1 tvrdých tobolek, ve vícečetném balení obsahujícím 3 balení po 60x1 tvrdých tobolek (180 tvrdých tobolek) nebo ve vícečetném balení obsahujícím 2 balení 50x1 tvrdých tobolek (100 tvrdých tobolek) v hliníku blistry. dělitelné jednotkovou dávkou.
Pradaxa 150mg tvrdé tobolky jsou také k dispozici v baleních obsahujících 60x1 tvrdých tobolek v bílých perforovaných jednodávkových hliníkových blistrech.
Tvrdé tobolky Pradaxa 150 mg jsou také k dispozici v polypropylenových (plastových) lahvích obsahujících 60 tvrdých tobolek.
Na trhu nemusí být všechny velikosti balení
Zdroj příbalové informace: AIFA (Italská agentura pro léčivé přípravky). Obsah zveřejněný v lednu 2016. Přítomné informace nemusí být aktuální.
Chcete-li mít přístup k nejaktuálnější verzi, doporučujeme navštívit webovou stránku AIFA (Italská agentura pro léčivé přípravky). Prohlášení a užitečné informace.
01.0 NÁZEV LÉČIVÉHO PŘÍPRAVKU
PRADAXA 110 MG TVRDÉ Kapsle
02.0 KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje 110 mg dabigatran -etexilátu (jako mesylátu).
Pomocné látky se známými účinky:
Jedna tvrdá tobolka obsahuje 3 mcg oranžové žluti (E110).
Úplný seznam pomocných látek viz bod 6.1.
03.0 LÉKOVÁ FORMA
Tvrdá tobolka.
Kapsle s neprůhledným světle modrým víčkem a neprůhledným krémovým tělem velikosti 1 naplněné nažloutlými peletami. Logo Boehringer Ingelheim je vytištěno na hlavě, „R110“ na těle.
04.0 KLINICKÉ INFORMACE
04.1 Terapeutické indikace
Primární prevence tromboembolických epizod u dospělých pacientů podstupujících elektivní totální operaci náhrady kyčle nebo kolena.
Prevence cévní mozkové příhody a systémové embolie u dospělých pacientů s nevalvulární fibrilací síní (NVAF) s jedním nebo více rizikovými faktory, jako je předchozí mrtvice nebo přechodný ischemický záchvat (TIA); věk ≥ 75 let; srdeční selhání (třída NYHA ≥ II); diabetes mellitus; hypertenze.
Léčba hluboké žilní trombózy (DVT) a plicní embolie (PE) a prevence recidivy DVT a PE u dospělých.
04.2 Dávkování a způsob podání
Dávkování
Primární prevence epizod žilního tromboembolismu v ortopedické chirurgii
Pacienti podstupující elektivní operaci kolenní náhrady
Doporučená dávka přípravku Pradaxa je 220 mg jednou denně, užívaná jako 2 kapsle po 110 mg. Léčba by měla být zahájena orálně během 1-4 hodin po dokončení chirurgického zákroku jednou 110mg tobolkou a pokračovat od druhého dne 2 kapslemi jednou denně po dobu celkem 10 dnů.
Pacienti podstupující elektivní operaci náhrady kyčle
Doporučená dávka přípravku Pradaxa je 220 mg jednou denně, užívaná jako 2 kapsle po 110 mg. Léčba by měla být zahájena orálně během 1-4 hodin po dokončení chirurgického zákroku s 110 mg tobolkou a pokračovat od druhého dne 2 tobolkami jednou denně po dobu celkem 28-35 dnů.
Pro následující skupiny je doporučená denní dávka přípravku Pradaxa 150 mg jednou denně, užívaná jako 2 tobolky po 75 mg.
Léčba by měla být zahájena orálně během 1-4 hodin po dokončení chirurgického zákroku s jednou 75mg tobolkou a pokračovat od dalšího dne 2 tobolkami jednou denně po dobu celkem 10 dnů (operace kolenní náhrady) nebo 28 dnů. -35 dní (náhrada kyčle) chirurgická operace):
• Pacienti se středně těžkou poruchou funkce ledvin (clearance kreatininu, CrCL 30–50 ml / min [viz Porucha funkce ledvin (primární prevence epizod žilního tromboembolismu při ortopedické chirurgii)]
• Pacienti souběžně užívající verapamil, amiodaron, chinidin [viz Souběžné podávání přípravku Pradaxa se slabými až středně silnými inhibitory P-glykoproteinu (P-gp), jako je amiodaron, chinidin nebo verapamil (primární prevence žilního tromboembolismu při ortopedické chirurgii)]
• Pacienti ve věku 75 let a starší [viz Starší pacienti (primární prevence epizod žilního tromboembolismu v ortopedické chirurgii)]
Pokud u obou intervencí není hemostáza normální, zahájení léčby by mělo být odloženo. Pokud léčba není zahájena v den operace, měla by být zahájena 2 tobolkami jednou denně.
Vyhodnocení funkce ledvin (primární prevence epizod žilního tromboembolismu v ortopedické chirurgii):
U všech pacientů:
• Renální funkce by měla být hodnocena výpočtem clearance kreatininu (CrCL) před zahájením léčby přípravkem Pradaxa, aby se vyloučili pacienti s těžkou poruchou funkce ledvin (tj. CrCL
• Renální funkce by měla být vyhodnocena také v případě podezření na sníženou funkci ledvin během léčby (např. Hypovolémie, dehydratace a v případě souběžného užívání některých léčivých přípravků).
Metoda používaná k odhadu funkce ledvin (CrCL v ml / min) během klinického vývoje přípravku Pradaxa byla metoda Cockgroft-Gault (viz bod 4.2 přípravku Pradaxa 75 mg).
Zvláštní populace
Porucha funkce ledvin (primární prevence epizod žilního tromboembolismu v ortopedické chirurgii)
Léčba přípravkem Pradaxa u pacientů s těžkou poruchou funkce ledvin (CrCLr
Klinické zkušenosti u pacientů se středně těžkou poruchou funkce ledvin (CrCL 30-50 ml / min) jsou omezené.Tito pacienti by měli být léčeni opatrně.Doporučená dávka je 150 mg jednou denně ve formě 2 75mg tobolek (viz body 4.4 a 5.1).
Souběžné užívání přípravku Pradaxa se slabými až středně silnými inhibitory P-glykoproteinu (P-gp), jako je amiodaron, chinidin nebo verapamil (primární prevence žilního tromboembolismu při ortopedické chirurgii)
U pacientů, kteří současně dostávají dabigatranetexilát a amiodaron, chinidin nebo verapamil, by měla být dávka přípravku Pradaxa snížena na 150 mg, užívaných jednou denně jako dvě 75mg tobolky (viz body 4.4 a 4.5). V tomto případě musí být přípravek Pradaxa a tyto léky užívány společně.
U pacientů se středně těžkou poruchou funkce ledvin, kteří jsou souběžně léčeni dabigatranetexilátem a verapamilem, by mělo být zváženo snížení dávky přípravku Pradaxa na 75 mg denně (viz body 4.4 a 4.5).
Starší osoby (primární prevence epizod žilního tromboembolie v ortopedické chirurgii)
Klinické zkušenosti jsou u starších pacientů (> 75 let) omezené.Tito pacienti by měli být léčeni opatrně.Doporučená dávka je 150 mg jednou denně ve formě dvou 75mg tobolek (viz body 4.4 a 5.1).
Vzhledem k tomu, že poškození funkce ledvin může být u starších osob (věk> 75 let) časté, funkce ledvin by měla být posouzena výpočtem CrCL před zahájením léčby přípravkem Pradaxa, aby se vyloučili pacienti s těžkou poruchou funkce ledvin (tj.
Porucha funkce jater (primární prevence epizod žilního tromboembolismu v ortopedické chirurgii)
Pacienti s jaterními enzymy zvýšenými nad dvojnásobek horní hranice normálu (ULN) byli vyloučeni z klinických studií hodnotících prevenci VTE po elektivní totální náhradě kyčle nebo kolena.Neexistují žádné zkušenosti s léčbou této subpopulace pacientů, a proto použití přípravku Pradaxa se v této populaci nedoporučuje (viz body 4.4 a 5.2). Je kontraindikováno v případě poškození jater nebo onemocnění jater, které může mít jakýkoli dopad na přežití (viz bod 4.3).
Hmotnost (primární prevence epizod žilního tromboembolie v ortopedické chirurgii)
Klinické zkušenosti s doporučenou dávkou u pacientů s tělesnou hmotností 110 kg jsou velmi omezené.Na základě klinických a kinetických údajů není nutná úprava dávky (viz bod 5.2), ale doporučuje se pečlivé klinické sledování (viz bod 4.4).
Pohlaví (primární prevence epizod žilního tromboembolie v ortopedické chirurgii)
Na základě dostupných klinických a kinetických údajů není nutná úprava dávky (viz bod 5.2).
Přepínání (primární prevence epizod žilního tromboembolie v ortopedické chirurgii)
Od léčby přípravkem Pradaxa k parenterálnímu antikoagulantu
Doporučuje se počkat 24 hodin po poslední dávce, než přejdete z přípravku Pradaxa na parenterální antikoagulant (viz bod 4.5).
Od parenterálních antikoagulancií po Pradaxu
Zadržte parenterální antikoagulant a dabigatranetexilát zahajte 0–2 hodiny před plánovaným podáním další dávky původní terapie nebo po přerušení v případě kontinuální léčby (např. Intravenózní nefrakcionovaný heparin (ENF)) (viz bod 4.5).
Dětská populace (primární prevence epizod žilního tromboembolie v ortopedické chirurgii)
Neexistuje žádné relevantní použití přípravku Pradaxa u pediatrické populace v indikaci: primární prevence epizod žilního tromboembolismu u pacientů podstupujících elektivní totální náhradu kyčelního kloubu nebo elektivní totální náhradu kolena.
Zmeškaná dávka (primární prevence epizod žilního tromboembolie v ortopedické chirurgii)
Doporučuje se, aby zbývající denní dávky dabigatran -etexilátu pokračovaly ve stejnou dobu následujícího dne.
Nezdvojnásobujte dávky, abyste nahradili zapomenutou dávku.
Dávkování (prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE)
Prevence cévní mozkové příhody a ES u dospělých pacientů s NSAID s jedním nebo více rizikovými faktory (prevence cévní mozkové příhody u pacientů s fibrilací síní)
Doporučená denní dávka přípravku Pradaxa je 300 mg užívaná jako jedna 150mg tobolka dvakrát denně. Terapie musí pokračovat dlouhodobě.
Léčba hluboké žilní trombózy (DVT) a plicní embolie (PE) a prevence recidivy DVT a PE u dospělých (DVT / PE)
Doporučená denní dávka přípravku Pradaxa je 300 mg užívaná jako jedna 150mg tobolka dvakrát denně po léčbě parenterálním antikoagulantem podávaným po dobu nejméně 5 dnů. Trvání léčby by mělo být stanoveno po „pečlivém zhodnocení přínosu terapie vůči riziku krvácení (viz bod 4.4). Volba krátkodobé terapie (nejméně 3 měsíce) by měla být založena na přechodných rizikových faktorech (např. nedávné chirurgické zákroky, traumata, imobilizace) a dlouhodobější operace s trvalými rizikovými faktory nebo idiopatickou DVT nebo PE.
Prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE
Pro následující skupiny je doporučená denní dávka přípravku Pradaxa 220 mg ve formě jedné 110mg tobolky dvakrát denně:
• Pacienti ve věku 80 let a starší
• Pacienti na souběžné léčbě verapamilem
U následujících skupin by měla být denní dávka přípravku Pradaxa 300 mg nebo 220 mg určena individuálně na základě posouzení rizika tromboembolie a rizika krvácení:
• Pacienti ve věku 75 až 80 let
• Pacienti se středně těžkou poruchou funkce ledvin
• Pacienti s gastritidou, ezofagitidou nebo gastroezofageálním refluxem
• Ostatní pacienti se zvýšeným rizikem krvácení
U DVT / PE je doporučení použít 220 mg přípravku Pradaxa užívaného jako jedna 110mg tobolka dvakrát denně založeno na farmakokinetických a farmakodynamických hodnoceních a v této klinické podskupině nebylo studováno.
Viz další informace v bodech 4.4, 4.5, 5.1 a 5.2 níže.
V případě nesnášenlivosti dabigatranu by pacienti měli být poučeni, aby neprodleně kontaktovali svého lékaře, který je může převést na přijatelné alternativní léčebné možnosti prevence cévní mozkové příhody a ES spojené s fibrilací síní nebo DVT / PE.
Starší pacienti (prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE)
Pacienti ve věku 75 až 80 let by měli být léčeni denní dávkou 300 mg užitou jako jedna 150mg tobolka dvakrát denně. Pokud je riziko tromboembolie nízké a riziko krvácení vysoké (individuálně) a podle uvážení lékaře, může být zvážena denní dávka 220 mg užívaná jako jedna 110mg tobolka dvakrát denně (viz bod 4.4).
Pacienti ve věku 80 let nebo starší by měli být léčeni denní dávkou 220 mg užívanou jako jedna 110mg tobolka dvakrát denně, kvůli zvýšenému riziku krvácení v této populaci.
Vzhledem k tomu, že poškození funkce ledvin může být u starších osob (věk> 75 let) časté, funkce ledvin by měla být posouzena výpočtem CrCL před zahájením léčby přípravkem Pradaxa, aby se vyloučili pacienti s těžkou poruchou funkce ledvin (tj.
Pacienti s rizikem krvácení (prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE)
Pacienti se zvýšeným rizikem krvácení (viz body 4.4, 4.5, 5.1 a 5.2) by měli podstoupit „pečlivé klinické sledování (hledat známky krvácení nebo anémie). Úpravu dávky je třeba rozhodnout podle uvážení lékaře.“, Po zhodnocení prospěch a potenciální riziko pro každého jednotlivého pacienta. Koagulační test (viz bod 4.4) může pomoci identifikovat pacienty se zvýšeným rizikem krvácení v důsledku „zvýšené expozice dabigatranu. Pokud je u pacientů s vysokým rizikem krvácení zjištěna nadměrná expozice dabigatranu, dávka 220 mg užívaná jako jedna 110 mg kapsle dvakrát denně. Pokud dojde ke klinicky relevantnímu krvácení, léčba by měla být ukončena.
U osob s gastritidou, ezofagitidou nebo refluxem jícnu lze uvažovat o dávce 220 mg užívané jako jedna 110mg tobolka dvakrát denně kvůli vysokému riziku závažného gastrointestinálního krvácení (viz bod 4.4).
Hodnocení funkce ledvin (prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE):
U všech pacientů:
• Renální funkce by měla být hodnocena výpočtem clearance kreatininu (CrCL) před zahájením léčby přípravkem Pradaxa, aby se vyloučili pacienti s těžkou poruchou funkce ledvin (tj. CrCL
• Renální funkce by měla být vyhodnocena také v případě podezření na sníženou funkci ledvin během léčby (např. Hypovolémie, dehydratace a v případě souběžného užívání některých léčivých přípravků).
Další požadavky pro pacienty s mírnou až středně těžkou poruchou funkce ledvin a pro pacienty starší 75 let:
• Renální funkce by měla být během léčby přípravkem Pradaxa hodnocena nejméně jednou ročně nebo častěji podle potřeby v určitých klinických situacích, kdy existuje podezření na sníženou nebo zhoršující se funkci ledvin (např. Hypovolémie, dehydratace a v případě souběžného užívání některých léků).
Metoda používaná k odhadu funkce ledvin (CrCL v ml / min) během klinického vývoje přípravku Pradaxa byla metoda Cockgroft-Gault (viz bod 4.2 přípravku Pradaxa 75 mg).
Porucha funkce ledvin (prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE)
Léčba přípravkem Pradaxa u pacientů s těžkou poruchou funkce ledvin (CrCLr
U pacientů s mírnou poruchou funkce ledvin (CrCL 50 ≤ 80 ml / min) není nutná úprava dávky. I u pacientů se středně těžkou poruchou funkce ledvin (CrCL 30-50 ml / min) je doporučená dávka přípravku Pradaxa 300 mg užitá jako jedna 150mg tobolka dvakrát denně. U pacientů s vysokým rizikem krvácení by však mělo být zváženo snížení dávky přípravku Pradaxa na 220 mg užívaných jako jedna 110mg tobolka dvakrát denně (viz body 4.4 a 5.2). U pacientů s poruchou funkce ledvin se doporučuje pečlivé klinické sledování.
Souběžné užívání přípravku Pradaxa se slabými až středně silnými inhibitory P-glykoproteinu (P-gp), jako je amiodaron, chinidin nebo verapamil (prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE)
Při souběžném podávání s amiodaronem nebo chinidinem není nutná úprava dávky (viz body 4.4, 4.5 a 5.2).
U pacientů užívajících dabigatran -etexilát i verapamil by měla být dávka dabigatranu snížena na 220 mg užívaných jako jedna 110mg tobolka dvakrát denně (viz body 4.4 a 4.5). V tomto případě musí být přípravek Pradaxa a verapamil užívány společně.
Hmotnost (prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE)
Na základě dostupných klinických a kinetických údajů není nutná úprava dávky (viz bod 5.2), ale u pacientů s tělesnou hmotností se doporučuje pečlivé klinické sledování.
Pohlaví (prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE)
Na základě dostupných klinických a kinetických údajů není nutná úprava dávky (viz bod 5.2).
Porucha funkce jater (prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE)
Pacienti s jaterními enzymy zvýšenými nad dvojnásobek horní hranice normálu (ULN) byli z pivotních klinických studií vyloučeni. S touto subpopulací pacientů nejsou žádné zkušenosti s léčbou, a proto se použití přípravku Pradaxa v této populaci nedoporučuje (viz body 4.4 a 5.2). Je kontraindikován v případě poškození jater nebo onemocnění jater, které může mít jakýkoli dopad na přežití (viz bod 4.3).
Přepínání (prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE)
Od léčby přípravkem Pradaxa k parenterálnímu antikoagulantu
Doporučuje se počkat 12 hodin po poslední dávce, než přejdete z dabigatranetexilátu na parenterální antikoagulant (viz bod 4.5).
Od parenterálních antikoagulancií po Pradaxu
Zadržte parenterální antikoagulancium a zahajte dabigatran-etexilát 0-2 hodiny před plánovanou další dávkou původní terapie nebo po přerušení v případě kontinuální léčby (např. Intravenózní nefrakcionovaný heparin (ENF)) (viz bod 4.5).
Od Pradaxy po antagonisty vitaminu K (AVK)
Zahájení terapie VKA by mělo být stanoveno na základě CRC podle následujících indikací:
• CrCL ≥ 50 ml / min, zahajte VKA 3 dny před vysazením dabigatran -etexilátu
• CrCL ≥ 30-
Protože Pradaxa může zvýšit hodnotu INR, bude lépe odrážet účinek AVK až po uplynutí alespoň 2 dnů od ukončení léčby přípravkem Pradaxa. Do té doby by měly být hodnoty INR interpretovány opatrně.
Od AVK po Pradaxu
AVK by měla být vysazena. Dabigatran -etexilát lze podat, jakmile je stanoven mezinárodní normalizovaný poměr (INR).
Kardioverze (prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE)
Pacienti podstupující kardioverzi mohou pokračovat v léčbě dabigatranem.
Pediatrická populace (prevence cévní mozkové příhody u pacientů s fibrilací síní)
Neexistuje žádné relevantní použití přípravku Pradaxa u pediatrické populace v indikaci: prevence mrtvice a systémové embolie u pacientů s NVAF.
Pediatrická populace (DVT / PE)
Bezpečnost a účinnost přípravku Pradaxa u dětí od narození do 18 let nebyla dosud stanovena. V současné době jsou dostupná data popsána v bodech 4.8 a 5.1, ale nelze stanovit doporučení ohledně dávkování.
Zmeškaná dávka (prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE)
Vynechanou dávku dabigatran -etexilátu je možné užít až 6 hodin před užitím další dávky.Potom je třeba vynechanou dávku vynechat.
Nezdvojnásobujte dávky, abyste nahradili zapomenutou dávku.
Způsob podání (primární prevence epizod žilního tromboembolie v ortopedické chirurgii, prevence cévní mozkové příhody u pacientů s fibrilací síní, DVT / PE)
Přípravek Pradaxa lze užívat s jídlem nebo bez jídla. Pradaxa by měla být spolknuta celá se sklenicí vody, aby se usnadnilo uvolňování žaludku.
Pacienti by měli být poučeni, aby neotvírali tobolky, protože to může vést ke zvýšenému riziku krvácení (viz body 5.2 a 6.6).
04.3 Kontraindikace
• Přecitlivělost na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1
• Pacienti s těžkou poruchou funkce ledvin (CrCL
• Klinicky významné aktivní krvácení
• Zranění nebo stavy, pokud jsou považovány za významný rizikový faktor pro závažné krvácení. Mohou zahrnovat aktuální nebo nedávný gastrointestinální vřed, vysoké riziko krvácivých novotvarů, nedávné poranění mozku nebo páteře, nedávné operace mozku, páteře nebo oka, nedávné nitrolební krvácení, známé nebo suspektní jícnové varixy, arteriovenózní malformace, cévní aneuryzma nebo velké intraspinální nebo intracerebrální vaskulární abnormality
• Souběžná léčba jakýmkoli jiným antikoagulantem, jako je nefrakcionovaný heparin (ENF), nízkomolekulární heparin (enoxaparin, dalteparin atd.), Deriváty heparinu (fondaparinux atd.), Perorální antikoagulancia (warfarin, rivaroxaban, apixaban atd.), S výjimkou specifických okolnosti změny antikoagulační terapie (viz bod 4.2) nebo pokud je ENF podáván v dávkách nezbytných pro zachování patentu centrálního žilního nebo arteriálního katétru (viz bod 4.5)
• Porucha funkce jater nebo onemocnění jater, které mohou mít jakýkoli dopad na přežití
• Souběžná léčba systémovým ketokonazolem, cyklosporinem, itrakonazolem a dronedaronem (viz bod 4.5)
• Protézy srdeční chlopně vyžadující antikoagulační léčbu (viz bod 5.1).
04.4 Zvláštní upozornění a vhodná opatření pro použití
Porucha funkce jater
Pacienti se zvýšenými jaterními enzymy nad dvojnásobek horní hranice normálu byli z pivotních klinických studií vyloučeni. S touto subpopulací pacientů nejsou žádné zkušenosti s léčbou, a proto se použití přípravku Pradaxa v této populaci nedoporučuje.
Riziko krvácení
Dabigatran -etexilát by měl být používán s opatrností v podmínkách zvýšeného rizika krvácení a v situacích zahrnujících souběžné použití s látkami, které mění hemostázu inhibicí agregace krevních destiček. Během léčby může dojít ke krvácení. S dabigatran -etexilátem. Nevysvětlitelný pokles hemoglobinu a / nebo hematokrit nebo krevní tlak by měly vyvolat hledání místa krvácení.
Faktory, jako je snížená funkce ledvin (30-50 ml / min CrCl), věk ≥ 75 let, nízké plazmatické hladiny dabigatranu (viz body 4.2, 4.5 a 5.2).
Současné užívání tikagreloru zvyšuje expozici dabigatranu a může vést k farmakodynamickým interakcím, které mohou mít za následek zvýšené riziko krvácení (viz bod 4.5).
Ve studii prevence cévní mozkové příhody a SE u dospělých pacientů s NVAF byl dabigatran -etexilát spojen s vyšším výskytem závažného gastrointestinálního (GI) krvácení, které bylo statisticky významné pro dabigatran -etexilát 150 mg dvakrát denně. Toto zvýšené riziko bylo pozorováno u starších osob (≥ 75 let). Riziko gastrointestinálního krvácení zvyšuje také používání kyseliny acetylsalicylové (ASA), klopidogrelu nebo nesteroidních protizánětlivých léků (NSAID), jakož i přítomnost ezofagitidy, gastritidy nebo gastroezofageálního refluxu. U těchto pacientů s fibrilací síní by měla být zvážena dávka 220 mg užitá jako jedna 110mg tobolka dvakrát denně a mělo by být dodrženo dávkování doporučené v bodě 4.2. K prevenci gastrointestinálního krvácení lze zvážit podání PPI.
Riziko krvácení může být zvýšeno u pacientů souběžně léčených selektivními inhibitory zpětného vychytávání serotoninu (SSRI) nebo selektivními inhibitory zpětného vychytávání serotoninu a norepinefrinu (SNRI) (viz bod 4.5).
Během léčby se doporučuje pečlivé klinické sledování (hledání příznaků krvácení nebo anémie), zvláště pokud jsou rizikové faktory kombinovány (viz bod 5.1).
Tabulka 1 shrnuje faktory, které mohou zvýšit riziko krvácení. Viz také kontraindikace v bodě 4.3.
Tabulka 1: Faktory, které mohou zvýšit riziko krvácení
Přítomnost lézí, stavů, postupů a / nebo léčby léky (jako jsou NSAID, protidestičková činidla, SSRI a SNRI, viz bod 4.5), které významně zvyšují riziko závažného krvácení, vyžaduje pečlivé „vyhodnocení přínosu a rizika“. být podáván pouze tehdy, pokud přínos převáží riziko krvácení.
Pradaxa běžně nevyžaduje rutinní sledování parametrů koagulace. Posouzení antikoagulačního účinku souvisejícího s dabigatranem však může být užitečné k zamezení nadměrně vysoké expozice dabigatranu za přítomnosti dalších rizikových faktorů. Test INR není spolehlivý u pacientů léčených přípravkem Pradaxa a bylo hlášeno falešně pozitivní zvýšení INR. Test INR by proto neměl být prováděn. Zředěný plazmatický trombinový čas (dTT), ekarinový čas (ECT), aktivovaný parciální tromboplastinový čas (aPTT) mohou poskytnout užitečné informace, ale testy nejsou standardizované a výsledky by měly být interpretovány s opatrností (viz bod 5.1).
Tabulka 2 ukazuje mezní hodnoty v době koagulačních testů, které mohou být spojeny se zvýšeným rizikem krvácení (viz bod 5.1).
Tabulka 2: Mezní hodnoty prahové hodnoty v době minimální doby koagulačních testů, které mohou být spojeny se zvýšeným rizikem krvácení
Pacienti, u nichž se rozvine akutní selhání ledvin, by měli přestat užívat přípravek Pradaxa (viz bod 4.3).
Údaje o hmotnostních pacientech
Pokud dojde k závažnému krvácení, léčba by měla být zastavena a měl by být vyšetřen zdroj krvácení (viz bod 4.9).
Léčivé přípravky, které mohou zvýšit riziko krvácení, by neměly být podávány současně nebo by měly být s přípravkem Pradaxa podávány s opatrností (viz bod 4.5).
Použití fibrinolytických léčiv k léčbě akutní ischemické cévní mozkové příhody
O použití fibrinolytických léčivých přípravků k léčbě akutní ischemické cévní mozkové příhody lze uvažovat, pokud má pacient dTT, ECT nebo aPTT pod horní hranicí normálu podle místního referenčního rozmezí.
Interakce s induktory P-gp
Při současném podávání induktorů P-gp (jako je rifampicin, třezalka (Hypericum perforatum), karbamazepin nebo fenytoin) lze očekávat snížení plazmatických koncentrací dabigatranu, kterým je třeba se vyvarovat (viz body 4.5 a 5.2).
Chirurgie a intervence
Pacienti na dabigatranetexilátu, kteří podstupují chirurgický zákrok nebo invazivní procedury, mají zvýšené riziko krvácení. Chirurgické zákroky proto mohou vyžadovat dočasné přerušení léčby.
Opatrnosti a monitorování antikoagulační aktivity se doporučuje, když je léčba dočasně pozastavena z důvodu chirurgického zákroku. Clearance dabigatranu u pacientů s renální insuficiencí může trvat déle (viz bod 5.2). To je třeba vyhodnotit před každým postupem. V takových případech koagulační test (viz body 4.4 a 5.1) může pomoci určit, zda je hemostáza stále narušena.
Předoperační fáze
Tabulka 3 shrnuje pravidla pro stažení před invazivními nebo chirurgickými zákroky.
Tabulka 3: Pravidla pro výběr před invazivními nebo chirurgickými postupy
Pokud je vyžadována naléhavá akce, měl by být dabigatran -etexilát dočasně pozastaven. Operace / intervence, pokud je to možné, by měla být odložena nejméně na 12 hodin po poslední užité dávce. Pokud nelze operaci odložit, může dojít ke zvýšenému riziku krvácení.Toto riziko krvácení je třeba zvážit s ohledem na naléhavost chirurgického zákroku (kardioverze viz bod 4.2).
Spinální anestezie / epidurální anestezie / lumbální punkce
Procedury, jako je spinální anestézie, vyžadují normální hemostatické funkce.
Riziko spinálního nebo epidurálního hematomu může být zvýšeno v případě traumatické nebo opakované punkce a dlouhodobým používáním epidurálních katetrů.Po odstranění katétru by měl před podáním první dávky dabigatranetexilátu uplynout alespoň 2hodinový interval. Tito pacienti vyžadují časté sledování neurologických příznaků a symptomů spinálního nebo epidurálního hematomu.
Pooperační fáze
Podávání dabigatranetexilátu by mělo být obnoveno co nejdříve po invazivním zákroku nebo chirurgickém zákroku za předpokladu, že bylo prokázáno, že klinická situace umožňuje adekvátní hemostázu.
Pacienti s vysokým rizikem krvácení nebo pacienti s rizikem nadměrné expozice, zejména pacienti se středně těžkou poruchou funkce ledvin (CrCL 30-50 ml / min), by měli být léčeni opatrně (viz body 4.4 a 5.1).
Pacienti s vysokým rizikem úmrtnosti v důsledku chirurgického zákroku as vnitřními rizikovými faktory pro tromboembolické příhody
U těchto pacientů jsou k dispozici omezené údaje o účinnosti a bezpečnosti dabigatranu, a proto by s nimi mělo být zacházeno opatrně.
Operace zlomeniny kyčle
Nejsou k dispozici žádné údaje o použití přípravku Pradaxa u pacientů podstupujících operaci zlomeniny kyčle. Léčba se proto nedoporučuje.
Infarkt myokardu (prevence mrtvice u pacientů s fibrilací síní)
Ve studii fáze III RE-LY (viz bod 5.1) byl celkový výskyt infarktu myokardu (MI) 0,82, 0,81 a 0,64% / rok pro dabigatranetexilát 110 mg dvakrát denně, dabigatranetexilát 150 mg dvakrát denně a warfarin s zvýšení relativního rizika pro dabigatran o 29% a 27% ve srovnání s warfarinem. Bez ohledu na následnou terapii bylo největší absolutní riziko MI pozorováno v následujících podskupinách s podobným relativním rizikem: pacienti s předchozím IM, pacienti ≥ 65 let věk s diabetem nebo ischemickou chorobou srdeční, pacienti s ejekční frakcí levé komory
Infarkt myokardu (DVT / PE)
Ve třech aktivně kontrolovaných klinických studiích byl u pacientů léčených dabigatran-etexilátem hlášen vyšší výskyt MI než u pacientů léčených warfarinem: 0,4% vs. 0,2%, v krátkodobém RE-COVER a RE-COVER II; a 0,8% oproti 0,1% v dlouhodobé studii RE-MEDY. V této studii byl nárůst statisticky významný (p = 0,022).
Ve studii RE-SONATE, která srovnávala dabigatranetexilát s placebem, byla incidence MI 0,1% u pacientů užívajících dabigatranetexilát a 0,2% u pacientů užívajících placebo.
Pacienti s aktivní rakovinou (DVT / PE)
Účinnost a bezpečnost nebyla u DVT / PE stanovena u pacientů s existujícím nádorovým onemocněním.
Barviva
Tvrdé tobolky Pradaxa obsahují barvivo oranžové žluté (E110), které může způsobit alergické reakce.
04.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Antikoagulancia a protidestičková činidla
S následujícími léčbami nejsou žádné nebo jen omezené zkušenosti, které by mohly zvýšit riziko krvácení při současném použití s přípravkem Pradaxa: antikoagulancia jako nefrakcionovaný heparin (ENF), nízkomolekulární heparin (LMWH) a deriváty heparinu (fondaparinux, desirudin), trombolytické léčivé přípravky přípravky a antagonisté vitaminu K, rivaroxaban nebo jiná perorální antikoagulancia (viz bod 4.3) a protidestičková činidla, jako jsou antagonisté receptoru GPIIb / IIIa, tiklopidin, prasugrel, tikagrelor, dextran a sulfinpyrazon (viz bod 4.4).
Z omezených údajů shromážděných ve studii fáze III RE-LY u pacientů s fibrilací síní bylo pozorováno, že souběžné užívání jiných perorálních nebo parenterálních antikoagulancií zvyšuje výskyt závažného krvácení přibližně 2,5krát, a to jak u dabigatranetexilátu, tak u warfarinu. , zvláště při přechodu z jednoho antikoagulancia na jiné (viz bod 4.3).
ENF lze podávat v dávkách nezbytných pro zachování patentu na centrální žilní nebo arteriální katetr (viz bod 4.3).
Clopidogrel a ASA: Z údajů shromážděných ve studii fáze III RE-LY (viz bod 5.1) bylo pozorováno, že současné užívání protidestičkových látek, ASA nebo klopidogrelu, přibližně zdvojnásobuje výskyt závažného krvácení jak u dabigatranetexilátu, tak u warfarinu (viz bod 4.4).
Clopidogrel: Ve studii fáze I u zdravých mladých dobrovolníků mužského pohlaví nevedlo současné podávání dabigatranetexilátu a klopidogrelu k dalšímu prodloužení kapilárního krvácení ve srovnání se samotným klopidogrelem. Kromě toho „AUC ?, Ss a Cmax, ss a koagulační opatření pro účinek dabigatranu nebo inhibice agregace krevních destiček jako měřítka účinku klopidogrelu“ zůstaly v podstatě nezměněny při srovnání kombinované léčby a příslušných mono- nasycovací dávka 300 mg nebo 600 mg klopidogrelu, AUC, ss a Cmax, ss dabigatranu byly zvýšeny přibližně o 30-40% (viz bod 4.4) (viz také níže uvedený pododdíl na „ASA).
ASA: Účinek souběžného podávání dabigatranetexilátu a ASA na riziko krvácení byl studován u pacientů s fibrilací síní ve studii fáze II, ve které bylo aplikováno randomizované souběžné podávání ASA. Na základě analýzy ASA. Logistická regrese, současné podávání ASA a dabigatranetexilátu 150 mg dvakrát denně může zvýšit riziko jakéhokoli typu krvácení z 12% na 18% a 24% při 81 mg respektive 325 mg ASA (viz bod 4.4).
NSAID: Ukázalo se, že NSAID podávaná jako krátkodobě působící analgetika v perioperačním období nejsou spojena se zvýšeným rizikem krvácení v kombinaci s dabigatran etexilátem. Chronické používání NSAID zvýšilo riziko krvácení přibližně o 50%. U obou dabigatran etexilátu a warfarin. Proto je vzhledem k riziku krvácení, zvláště u NSAID s poločasem eliminace> 12 hodin, doporučeno pečlivé sledování příznaků krvácení (viz bod 4.4).
LMWH: Souběžné užívání LMWH, jako je enoxaparin a dabigatran etexilát, nebylo konkrétně hodnoceno. Po přechodu z 3denní léčby enoxaparinem 40 mg podávaným jednou denně sc cestou, 24 hodin po podání LMWH. dávka enoxaparinu, expozice dabigatranu byla o něco nižší než po podání samotného dabigatran-etexilátu (jednorázová dávka 220 mg). Po podání dabigatran-etexilátu, kterému předcházela předběžná léčba enoxaparinem, byla pozorována větší aktivita anti-FXa / FIIa před léčbou dabigatranem samotný etexilát. Předpokládá se, že je to způsobeno hybným účinkem léčby enoxaparinem a není považováno za klinicky relevantní. Výsledky dalších testů antikoagulační aktivity související s dabigatranem nebyly významně upraveny před léčbou enoxaparinem.
Interakce související s metabolickým profilem dabigatranetexilátu a dabigatranu
Dabigatranetexilát a dabigatran nejsou metabolizovány systémem cytochromu P450 a nemají žádný účinek. in vitro na enzymy lidského cytochromu P450. Interakce se souvisejícími léčivými přípravky a dabigatranem se proto neočekávají.
Interakce transportéru
Inhibitory P-gp
Dabigatranetexilát je substrátem efluxního transportéru P-gp. Souběžné podávání s inhibitory P-gp (jako je amiodaron, verapamil, chinidin, ketokonazol, dronedaron, klarithromycin a tikagrelor) pravděpodobně povede ke zvýšení plazmatických koncentrací dabigatranu.
Pokud není výslovně uvedeno jinak, je při současném podávání dabigatranu se silnými inhibitory P-gp vyžadováno pečlivé klinické sledování (hledající známky krvácení nebo anémie). Koagulační test pomáhá identifikovat pacienty se zvýšeným rizikem krvácení v důsledku „zvýšené expozice dabigatranu (viz body 4.2, 4.4 a 5.1).
Následující silné inhibitory P-gp jsou kontraindikovány: systémově podávaný ketokonazol, cyklosporin, itrakonazol a dronedaron (viz bod 4.3) Souběžná léčba takrolimem se nedoporučuje Inhibitory P-gp ze slabých Mírné léky (např. Amiodaron, posakonazol, chinidin, verapamil a tikagrelor) by měly být používány s opatrností (viz body 4.2 a 4.4).
Ketokonazol: Ketokonazol po jednorázové perorální dávce 400 mg zvýšil celkovou AUC0-∞ a Cmax dabigatranu o 138%, respektive 135%, respektive o 153%, respektive 149%, po opakovaných perorálních dávkách 400 mg ketokonazolu jednou denně. Ketokonazol nezměnil čas do dosažení vrcholu, konečný poločas a střední dobu setrvání (viz bod 4.4) Souběžné použití se systémovým ketokonazolem je kontraindikováno (viz bod 4.3).
Dronedaron: Při současném podávání dabigatran-etexilátu a dronedaronu se celkové hodnoty AUC0-∞ a Cmax dabigatranu zvýšily přibližně 2,4násobně a 2,3násobně (+ 136% a 125%), v uvedeném pořadí, po více dávkách 400. mg dronedaronu bid, a přibližně 2,1násobek a 1,9násobek (+ 114% a 87%), v uvedeném pořadí, po jednorázové dávce 400 mg Dronedaron neovlivnil konečný poločas a renální clearance dabigatranu. dávky dronedaronu byly podány 2 hodiny po dabigatranetexilátu, zvýšení AUC0-d dabigatranu bylo 1,3násobné, respektive 1,6násobné. Souběžná léčba dronedaronem je kontraindikována.
Amiodaron: Když byl přípravek Pradaxa podáván současně s jednou perorální dávkou 600 mg amiodaronu, množství a rychlost absorpce amiodaronu a jeho aktivního metabolitu DEA se v podstatě nezměnily. AUC a Cmax dabigatranu se zvýšily přibližně o 60%, respektive 50%. Mechanismus interakce není zcela objasněn. Vzhledem k dlouhému poločasu amiodaronu může potenciální léková interakce přetrvávat několik týdnů po vysazení amiodaronu (viz body 4.2 a 4.4).
U pacientů léčených k prevenci VTE po operaci náhrady kyčle nebo kolena by měla být dávka přípravku Pradaxa snížena na 150 mg užívaných jednou denně ve formě 2 x 75 mg tobolek, pokud jsou léčeni současně dabigatran -etexilátem a amiodaronem (viz bod 4.2). Při kombinaci dabigatran -etexilátu s amiodaronem se doporučuje pečlivé klinické sledování, zvláště pokud dojde ke krvácení, a s mimořádnou opatrností v případě pacientů s mírnou až středně těžkou poruchou funkce ledvin.
Chinidin: Chinidin byl podáván v dávkách 200 mg každé 2 hodiny až do celkové dávky 1 000 mg. Dabigatranetexilát byl podáván dvakrát denně po dobu 3 po sobě jdoucích dnů, třetí den s chinidinem nebo bez něj. AUC, ss a Cmax, ss dabigatranu byly zvýšeny v průměru o 53%, respektive 56%, při současném podávání chinidinu (viz body 4.2 a 4.4).
U pacientů léčených k prevenci VTE po operaci náhrady kyčle nebo kolena by měla být dávka přípravku Pradaxa snížena na 150 mg užívaných jednou denně ve formě 2 x 75 mg tobolek, pokud jsou léčeni současně s dabigatranetexilátem a chinidinem (viz bod 4.2). Při kombinaci dabigatran -etexilátu s chinidinem se doporučuje pečlivé klinické sledování, zvláště pokud dojde ke krvácení, a s mimořádnou opatrností v případě pacientů s mírnou až středně těžkou poruchou funkce ledvin.
Verapamil: Když byl dabigatranetexilát (150 mg) podáván současně s perorálním verapamilem, Cmax a AUC dabigatranu se zvyšovaly, ale velikost této změny se měnila v závislosti na době podání a formulaci verapamilu (viz body 4.2 a 4.4).
Maximální zvýšení expozice dabigatranu bylo pozorováno při první dávce přípravku verapamil s okamžitým uvolňováním podané jednu hodinu před podáním dabigatranetexilátu (zvýšení Cmax přibližně o 180% a AUC přibližně o 150%). Účinek se postupně snižoval podáním přípravku s prodlouženým uvolňováním (zvýšení Cmax přibližně o 90% a AUC přibližně o 70%) nebo podáním více dávek verapamilu (zvýšení Cmax přibližně o 60% a zvýšení AUC přibližně o 50%).
Při současném podávání dabigatranu s verapamilem je proto nutné pečlivé klinické sledování (hledající známky krvácení nebo anémie). U pacientů s normální funkcí ledvin po operaci náhrady kyčle nebo kolena léčených současně dabigatranetexilátem a verapamilem by měla být dávka přípravku Pradaxa snížena na 150 mg užívaných jako 2 x 75 mg tobolky jednou denně. U pacientů se středně těžkou poruchou funkce ledvin léčených současně dabigatranetexilátu a verapamilu je třeba zvážit snížení dávky přípravku Pradaxa na 75 mg denně (viz body 4.2 a 4.4).
U pacientů s NSAID léčených z důvodu mrtvice a prevence SE a u pacientů s DVT / PE léčených dabigatranetexilátem a verapamilem by měla být dávka přípravku Pradaxa snížena na 220 mg užívaných jako jedna 110mg tobolka dvakrát denně (viz bod 4.2).
Při kombinaci dabigatranetexilátu s verapamilem se doporučuje pečlivé klinické sledování, zvláště pokud dojde ke krvácení, a s mimořádnou opatrností v případě pacientů s mírnou až středně těžkou poruchou funkce ledvin.
Při podávání verapamilu 2 hodiny po užití dabigatranetexilátu nebyly pozorovány žádné významné interakce (přibližně 10% zvýšení Cmax a přibližně 20% zvýšení AUC). To je vysvětleno úplnou absorpcí dabigatranu po 2 hodinách (viz bod 4.4).
Klarithromycin: Když byl klarithromycin (500 mg dvakrát denně) podáván v kombinaci s dabigatran -etexilátem zdravým dobrovolníkům, bylo pozorováno zvýšení AUC přibližně o 19% a Cmax přibližně o 15% bez vlivu na klinickou bezpečnost. dabigatranu, nelze klinicky významnou interakci vyloučit při kombinaci s klarithromycinem. Proto je třeba pečlivě kombinovat dabigatran -etexilát s klarithromycinem a zvláště v případě krvácení, zvláště u pacientů s mírnou až středně těžkou poruchou funkce ledvin.
Ticagrelor: Když byla podána jedna 75mg dávka dabigatranetexilátu souběžně se 180mg počáteční dávkou ticagreloru, AUC a Cmax dabigatranu se zvýšily o 1,73, respektive 1,95násobně (+73 % a 95 %). Po vícenásobném podání 90 mg tikagreloru nabízí zvýšení expozice dabigatranu 1,56, respektive 1,46násobek (+ 56% a 46%) pro AUC, respektive Cmax.
Souběžné podávání počáteční dávky 180 mg tikagreloru a 110 mg dabigatran-etexilátu (v ustáleném stavu) zvýšilo AUC?, Ss a Cmax, ss dabigatranu 1,49násobně a 1,65násobně (+49% a 65 %), ve srovnání s podáváním samotného dabigatranetexilátu. Když byla počáteční dávka 180 mg tikagreloru podána 2 hodiny po podání dabigatranetexilátu 110 mg (ustálený stav), zvýšení AUC?, Ss a Cmax, ss dabigatranu bylo sníženo na 1,27násobek, respektive 1,23krát (+ 27% a 23%), ve srovnání s podáváním samotného dabigatran -etexilátu. Toto střídavé podávání je doporučeno pro zahájení léčby tikagrelorem s počáteční dávkou.
Souběžné podávání 90 mg tikagreloru BID (udržovací dávka) se 110 mg dabigatran-etexilátu zvýšilo upravenou AUC ?, Ss a Cmax, ss dabigatranu o 1,26násobek, respektive 1,29násobek, ve srovnání s podáním samotného dabigatranetexilátu.
Následující silné inhibitory P-gp nebyly studovány klinicky, ale na základě údajů in vitro očekává se účinek podobný účinku ketokonazolu:
Itrakonazol a cyklosporin, které jsou kontraindikovány (viz bod 4.3).
In vitro bylo prokázáno, že takrolimus má podobný inhibiční účinek na P-gp, jaký byl pozorován u itrakonazolu a cyklosporinu. Dabigatran -etexilát nebyl klinicky studován v kombinaci s takrolimem. Omezené klinické údaje dostupné s jiným substrátem P-gp (everolimus) však naznačují, že inhibice P-gp takrolimem je slabší než ta, která byla pozorována u silných inhibitorů P-gp. Na základě těchto údajů se souběžná léčba takrolimem nedoporučuje .
Posaconazole také částečně inhibuje P-gp, ale nebyl klinicky studován. Souběžné podávání přípravku Pradaxa a posaconazolu by mělo být prováděno opatrně.
Induktory P-gp
Souběžné podávání induktoru P-gp (jako je rifampicin, třezalka (Hypericum perforatum), karbamazepin nebo fenytoin) může snížit koncentrace dabigatranu a je třeba se mu vyhnout (viz body 4.4 a 5.2).
Rifampicin: Předběžné podávání induktoru rifampicinu v dávce 600 mg jednou denně po dobu 7 dnů snížilo celkový vrchol dabigatranu o 65,5% a celkovou expozici o 67%. Indukční účinek byl snížen, což vedlo k expozici dabigatranu blízké referenční hodnotě do sedmého dne po přerušení léčby rifampicinem. Po dalších 7 dnech nebylo pozorováno žádné zvýšení biologické dostupnosti.
Jiné léky ovlivňující P-gp
Inhibitory proteázy, jako je ritonavir a jeho kombinace s jinými inhibitory proteázy, ovlivňují P-gp (jak jako inhibitory, tak jako induktory). Protože nebyly studovány, nedoporučují se pro souběžnou léčbu přípravkem Pradaxa.
Substrát P-gp
Digoxin: Ve studii zahrnující 24 zdravých pacientů, kdy byla Pradaxa podávána současně s digoxinem, nebyly pozorovány žádné změny v digoxinu ani významné klinické změny v expozici dabigatranu.
Souběžné užívání selektivních inhibitorů zpětného vychytávání serotoninu (SSRI) nebo se selektivními inhibitory zpětného vychytávání serotoninu a norepinefrinu (SNRI)
SSRI a SNRI zvyšovaly riziko krvácení ve všech léčebných skupinách studie RE-LY.
žaludeční pH
Pantoprazol: Když byl přípravek Pradaxa podáván v kombinaci s pantoprazolem, bylo pozorováno přibližně 30% zmenšení plochy pod křivkou závislosti plazmatické koncentrace na čase dabigatranu. Byl pozorován pantoprazol a další inhibitory protonové pumpy (PPI) .podávání s Pradaxou v klinických studiích a nezdálo se, že by souběžná léčba s PPI snižovala účinnost přípravku Pradaxa.
Ranitidin: Podávání ranitidinu přípravkem Pradaxa nemá žádný klinicky relevantní účinek na absorpci dabigatranu.
04.6 Těhotenství a kojení
Ženy v plodném věku / mužská a ženská antikoncepce
Ženy ve fertilním věku by se měly během léčby dabigatranetexilátem vyhnout otěhotnění.
Těhotenství
O použití dabigatran -etexilátu u těhotných žen je k dispozici omezené množství údajů.
Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Potenciální riziko pro člověka není známo.
Přípravek Pradaxa by neměl být během těhotenství podáván, pokud to není nezbytně nutné.
Čas krmení
Nejsou k dispozici žádné klinické údaje o účincích dabigatranu na kojence.
Kojení by mělo být během léčby přípravkem Pradaxa přerušeno.
Plodnost
U lidí nejsou k dispozici žádné údaje.
Ve studiích na zvířatech byl při dávce 70 mg / kg (plazmatická expozice 5krát vyšší než u pacientů) pozorován účinek na ženskou plodnost ve formě snížené implantace a zvýšené ztráty před implantací. Nebyly pozorovány žádné jiné účinky na plodnost žen. Nebyl zjištěn žádný vliv na mužskou plodnost. Při dávkách toxických pro matku (plazmatická expozice 5 až 10krát vyšší než u pacientů) byla u potkanů a králíků pozorována snížená tělesná hmotnost plodu a embryofetální životaschopnost se zvýšenými změnami plodu. V prenatálních a postnatálních studiích byla pozorována zvýšená úmrtnost plodu při dávkách toxických pro matky (dávka odpovídající 4krát vyšší plazmatické expozici než u pacientů).
04.7 Účinky na schopnost řídit a obsluhovat stroje
Pradaxa nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
04.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Celkem 10 795 pacientů bylo léčeno v 6 aktivně kontrolovaných studiích prevence VTE alespoň jednou silou studovaného léčiva. Z těchto 6 684 pacientů bylo léčeno 150 mg nebo 220 mg přípravku Pradaxa denně.
V klíčové studii zkoumající prevenci cévní mozkové příhody a SE u pacientů s fibrilací síní bylo dabigatranetexilátem léčeno celkem 12 042 pacientů.Z těchto 6 059 bylo léčeno 150 mg dabigatranetexilátu dvakrát denně, zatímco 5 983 bylo léčeno dabigatranetexilátem. dostávali dávky 110 mg dvakrát denně.
Ve dvou aktivně kontrolovaných studiích léčby DVT / PE, RE-COVER a RE-COVER II, bylo do bezpečnostní analýzy dabigatranetexilátu zahrnuto celkem 2553 pacientů. Všichni pacienti dostávali dávky dabigatranetexilátu 150 mg dvakrát denně. u obou léčebných postupů se dabigatran -etexilát a warfarin počítají od prvního příjmu dabigatran -etexilátu nebo warfarinu po přerušení parenterální léčby (pouze období orální léčby). To zahrnuje všechny nežádoucí účinky, které se vyskytly během léčby dabigatranem. léčba warfarinem je zahrnuta, kromě těch, které nastaly během překrývání mezi warfarinem a parenterální terapií.
Ve studii aktivní prevence DVT / PE, RE-MEDY, a preventivní studii placebo / DVT / PE, RE-SONATE, bylo léčeno celkem 2 114 pacientů. Všichni pacienti dostávali dávky dabigatranetexilátu 150 mg dvakrát denně.
Celkem asi 9% pacientů léčených elektivní operací kyčle nebo kolena (krátkodobá léčba do 42 dnů), 22% pacientů s fibrilací síní léčených pro cévní mozkovou příhodu a prevenci SE (dlouhodobá léčba do 3 let), 14 Nežádoucí účinky se vyskytly u% pacientů léčených pro DVT / PE a 15% pacientů léčených pro prevenci DVT / PE.
Nejčastěji hlášeným nežádoucím účinkem je krvácení, ke kterému došlo celkem u přibližně 14% pacientů léčených krátkodobě pro elektivní náhradu kyčle nebo kolena, u 16,6% pacientů s dlouhodobou fibrilací síní. 14,4% pacientů léčených pro DVT / PE. Kromě toho došlo ke krvácení u 19,4% pacientů ve studii prevence DVT / PE RE-MEDY a u 10,5% pacientů ve studii prevence DVT / PE RE-SONATE.
Protože populace pacientů léčených ve třech indikacích nejsou srovnatelná a krvácivé příhody jsou rozděleny do různých tříd orgánových systémů (SOC), je souhrnný popis epizod závažného krvácení a krvácení jakéhokoli druhu rozdělen podle indikací.
Ačkoli se to v klinických studiích objevovalo jen zřídka, mohou nastat závažné nebo závažné krvácivé příhody, které mohou být bez ohledu na místo zdravotně postižené, život ohrožující nebo dokonce smrt.
Souhrnná tabulka nežádoucích účinků
Tabulka 4 ukazuje nežádoucí účinky identifikované ve studiích primární prevence VTE po chirurgické náhradě kyčle nebo kolena, ve studii prevence tromboembolické cévní mozkové příhody a ES u pacientů s fibrilací síní a ve studiích léčby a prevence DVT / PE nařízených SOC. a frekvence s použitím následující konvence: velmi časté (≥ 1/10); časté (≥ 1/100,
Tabulka 4: Nežádoucí účinky
Primární prevence epizod žilního tromboembolismu v ortopedické chirurgii
Krvácející
Tabulka 5 uvádí počet (%) pacientů s nežádoucími krvácivými reakcemi během období prevence VTE ve dvou klíčových klinických studiích podle dávky.
Tabulka 5: Počet (%) pacientů s nežádoucími krvácivými reakcemi
Definice nežádoucích účinků závažného krvácení ve studiích RE-NOVATE a RE-MODEL byly následující:
• smrtelné krvácení
• klinicky manifestní krvácení spojené s poklesem hemoglobinu o ≥ 20 g / l (což odpovídá 1,24 mmol / l), obojí překračuje očekávané hodnoty
• klinicky manifestní krvácení překračující očekávané a vyžadující transfuzi ≥ 2 jednotek erytrocytů nebo plné krve nad očekávané
• symptomatické retroperitoneální, intrakraniální, nitrooční nebo intraspinální krvácení
• krvácení, které si vyžádalo přerušení léčby
• krvácení, které si vyžádalo novou operaci.
Objektivní testování bylo vyžadováno pro retroperitoneální krvácení (ultrazvukové vyšetření nebo počítačová tomografie (CT)) a intraspinální krvácení (CT nebo zobrazování magnetickou rezonancí).
Prevence mrtvice a ES u dospělých pacientů s NSAID s jedním nebo více rizikovými faktory
Krvácející
Tabulka 6 ukazuje hlavní krvácivé příhody zjištěné v pivotní studii hodnotící prevenci cévní mozkové příhody a SE u pacientů s fibrilací síní.
Tabulka 6: Události krvácení ve studii hodnotící mrtvici a prevenci SE u pacientů s atriální fibrilací
Velké krvácení bylo definováno splněním jednoho nebo více z následujících kritérií:
Krvácení spojené se snížením hemoglobinu nejméně o 20 g / l nebo vyžadující transfuzi alespoň 2 jednotek červených krvinek nebo plné krve.
Symptomatické krvácení v kritické „oblasti nebo orgánu: nitrooční, intrakraniální, intraspinální nebo intramuskulární s kompartmentovým syndromem, retroperitoneální krvácení, intraartikulární krvácení nebo perikardiální krvácení.
Velké krvácení bylo klasifikováno jako život ohrožující, pokud splňovalo jedno nebo více z následujících kritérií:
Smrtelné krvácení, symptomatické intrakraniální krvácení; snížení hemoglobinu nejméně o 50 g / l; transfuze alespoň 4 jednotek erytrocytů nebo plné krve; krvácení spojené s hypotenzí vyžadující použití inotropních léčivých přípravků podávaných intravenózně; krvácení, které si vyžádalo operaci.
Subjekty randomizované na dabigatranetexilát 110 mg dvakrát denně nebo 150 mg dvakrát denně měly výrazně nižší riziko život ohrožujícího krvácení a intrakraniálního krvácení než subjekty léčené warfarinem [p
Klinický přínos dabigatran-etexilátu z hlediska prevence cévní mozkové příhody a SE a snížení rizika ICH ve srovnání s warfarinem je zachován napříč jednotlivými podskupinami, např. Poškození ledvin, věk, souběžné užívání léčivých přípravků, jako jsou antiagregační látky nebo inhibitory P-gp. u některých podskupin pacientů je při léčbě antikoagulancii zvýšené riziko závažného krvácení, riziko nadměrného krvácení u dabigatranetexilátu je způsobeno gastrointestinálním krvácením, které se typicky vyskytuje během prvních 3–6 měsíců po zahájení terapie.
Léčba hluboké žilní trombózy (DVT) a plicní embolie (PE) a prevence recidivy DVT a PE u dospělých (léčba DVT / PE).
Tabulka 7 ukazuje krvácivé příhody v kombinované analýze pivotních studií RE-COVER a RE-COVER II, které testovaly léčbu hluboké žilní trombózy (DVT) a plicní embolie (PE). Primární cílové parametry závažného krvácení, závažné nebo klinicky relevantní krvácení a jakékoli krvácení bylo výrazně nižší než u warfarinu při nominální hladině alfa 5%.
Tabulka 7: Krvácení ve studiích RE-COVER a RE-COVER II, které testovaly léčbu hluboké žilní trombózy (DVT) a plicní embolie (PE)
Krvácení u obou léčeb se počítá od prvního příjmu dabigatranetexilátu nebo warfarinu po přerušení parenterální terapie (pouze období orální léčby). To zahrnuje všechny krvácivé příhody, ke kterým došlo během léčby dabigatranetexilátem. Zahrnuty jsou všechny krvácivé příhody, ke kterým došlo během léčby warfarinem, kromě těch, které se vyskytly během překrývání mezi warfarinem a parenterální terapií.
Definice závažných krvácivých příhod se řídila doporučeními Mezinárodní společnosti pro trombózu a hemostázu. Krvácející příhoda byla považována za závažnou, pokud splňovala alespoň jedno z následujících kritérií:
• Smrtelné krvácení
• Symptomatické krvácení v "kritické oblasti nebo orgánu, jako je intrakraniální, intraspinální, nitrooční, retroperitoneální, intraartikulární nebo perikardiální nebo intramuskulární s kompartmentovým syndromem. Aby bylo krvácení v" kritické oblasti nebo orgánu považováno za závažné, muselo být spojeno se situací. symptomatická klinika
• Krvácení, které způsobilo pokles hladiny hemoglobinu o 20 g / l (1,24 mmol / l) nebo více, nebo které vedlo k transfuzi 2 nebo více jednotek plné krve nebo červených krvinek.
Tabulka 8 ukazuje krvácivé příhody v pivotní studii RE-MEDY, která testovala prevenci hluboké žilní trombózy (DVT) a plicní embolie (PE). Některé krvácivé příhody (závažné, klinicky relevantní a jakékoli krvácení) byly významně menší než nominální alfa 5%u pacientů léčených dabigatranetexilátem ve srovnání s pacienty léčenými warfarinem.
Tabulka 8: Krvácení ve studii RE-MEDY, která testovala prevenci hluboké žilní trombózy (DVT) a plicní embolie (PE)
* HR nelze vyhodnotit, protože v jedné ze dvou kohort / ošetření nedošlo k žádné události
Definice závažných krvácivých příhod se řídila doporučeními Mezinárodní společnosti pro trombózu a hemostázu, jak je popsáno pro RECOVER a RECOVER II.
Tabulka 9 ukazuje události krvácení v pivotní studii RE-SONATE, která testovala prevenci hluboké žilní trombózy (DVT) a plicní embolie (PE). Procento kombinace závažného / klinicky významného krvácení a procento jakéhokoli krvácení bylo významně nižší při nominální hladině alfa 5% u pacientů užívajících placebo ve srovnání s pacienty užívajícími dabigatranetexilát.
Tabulka 9: Krvácení ve studii RE-SONATE, která testovala prevenci hluboké žilní trombózy (DVT) a plicní embolie (PE)
* HR nelze vyhodnotit, protože v jednom ze dvou ošetření nedošlo k žádné události
Definice závažných krvácivých příhod se řídila doporučeními Mezinárodní společnosti pro trombózu a hemostázu, jak je popsáno pro RECOVER a RECOVER II.
Infarkt
Prevence cévní mozkové příhody a systémové embolie u dospělých pacientů s nevalvulární fibrilací síní (NAVF) s jedním nebo více rizikovými faktory
Ve studii RE-LY byl ve srovnání s warfarinem roční výskyt infarktu myokardu u dabigatranetexilátu zvýšen z 0,64% (warfarin) na 0,82% (dabigatranetexilát 110 mg dvakrát denně) / 0,81% (dabigatranetexilát 150 mg dvakrát denně) ( viz bod 5.1).
Léčba hluboké žilní trombózy (DVT) a plicní embolie (PE) a prevence recidivy DVT a PE u dospělých (DVT / PE)
Ve třech aktivně kontrolovaných studiích byl u pacientů užívajících dabigatranetexilát zaznamenán vyšší výskyt MI než u pacientů užívajících warfarin: 0,4% oproti 0,2% v krátkodobém RE-COVER a RE-COVER II; a 0,8% oproti 0,1% v dlouhodobé studii RE-MEDY. V této studii byl nárůst statisticky významný (p = 0,022).
Ve studii RE-SONATE, která srovnávala dabigatranetexilát s placebem, byla incidence MI 0,1% u pacientů léčených dabigatranetexilátem a 0,2% u pacientů užívajících placebo (viz bod 4.4).
Pediatrická populace (DVT / PE)
V klinické studii 1160,88 došlo v období po léčbě u celkem 9 dospívajících pacientů (ve věku od 12 do bolestí břicha; bolest břicha) a 1 pacienta (11,1%) k závažné nepříbuzné události (recidivě VTE v noze), více než 3 dny po vysazení dabigatranetexilátu.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky vyskytující se po registraci léčivého přípravku je důležité, protože umožňuje průběžné sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení. "Adresa https: //www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
04.9 Předávkování
Dávky dabigatranetexilátu vyšší než doporučené vystavují pacienta zvýšenému riziku krvácení.
Pokud je podezření na předávkování, mohou koagulační testy pomoci určit riziko krvácení (viz body 4.4 a 5.1). Kvantitativní kalibrovaný test dTT nebo opakovaná měření dTT umožňují předpovědět, kdy bude dosaženo určitých hladin dabigatranu (viz bod 5.1), i když byla přijata jiná opatření, např. dialýza.
Nadměrná antikoagulační aktivita může vyžadovat přerušení léčby přípravkem Pradaxa. Pro dabigatran neexistuje žádné specifické antidotum. V případě krvácivých komplikací by měla být léčba přerušena a měla by být vyšetřena příčina krvácení. Vzhledem k tomu, že se dabigatran vylučuje převážně ledvinami, měla by být zachována adekvátní diuréza. Podle uvážení lékaře by měla být provedena vhodná podpůrná léčba, jako je chirurgická hemostáza a obnovení objemu krve.
Je možné zvážit koncentráty aktivovaného protrombinového komplexu (např. FEIBA) nebo rekombinantního faktoru VIIa nebo koncentráty koagulačních faktorů II, IX a X. Existují určité experimentální důkazy podporující úlohu těchto léčiv v boji proti antikoagulačnímu účinku dabigatranu, ale údaje o jejich užitečnosti v klinickém prostředí a také o možném riziku rebound tromboembolismu jsou velmi omezené. Koagulační testy se mohou stát nespolehlivými. Po podání léčiv kontrastujících s antikoagulačním účinkem. Při interpretaci výsledků těchto testů je nutná opatrnost. Podání koncentrátů krevních destiček by mělo být také zváženo, pokud dojde k trombocytopenii nebo byla použita dlouhodobě působící protidestičková činidla. Veškerá symptomatická léčba by měla být prováděna v souladu s rozhodnutím lékaře.
V závislosti na místní dostupnosti by v případě velkého krvácení měla být zvážena konzultace s odborníkem na koagulaci.
Protože je vazba na bílkoviny nízká, lze dabigatran dialyzovat; klinické zkušenosti prokazující užitečnost tohoto přístupu v klinických studiích jsou omezené (viz bod 5.2).
05.0 FARMAKOLOGICKÉ VLASTNOSTI
05.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antitrombotika, přímé inhibitory trombinu.
ATC kód: B01AE07.
Mechanismus účinku
Dabigatranetexilát je malé molekulární proléčivo, které nevykazuje žádnou farmakologickou aktivitu. Po perorálním podání je dabigatranetexilát rychle absorbován a přeměněn na dabigatran hydrolýzou katalyzovanou esterázou v plazmě a játrech. Dabigatran je účinný přímý, kompetitivní, reverzibilní inhibitor trombinu a je hlavní účinnou látkou v plazmě.
Jelikož trombin (serinová proteáza) umožňuje v koagulační kaskádě přeměnu fibrinogenu na fibrin, brání jeho inhibice tvorbě trombu. Dabigatran inhibuje volný trombin, trombin vázaný na fibrin a agregaci krevních destiček indukovanou trombinem.
Farmakodynamické účinky
Studie prováděné na zvířatech in-vivo A ex vivo prokázal antitrombotickou účinnost a antikoagulační aktivitu dabigatranu po intravenózním podání a dabigatranetexilátu po perorálním podání na různých zvířecích modelech trombózy.
Na základě údajů ze studií fáze II existuje jasná korelace mezi plazmatickou koncentrací dabigatranu a velikostí antikoagulačního účinku. Dabigatran prodlužuje trombinový čas (TT), ECT a aPTT.
Zkouška kalibrovaná pro dabigatran na zředěné plazmě pomocí trombinového času (dTT) poskytuje odhad plazmatické koncentrace dabigatranu, který lze porovnat s očekávanými plazmatickými koncentracemi dabigatranu.
ECT může poskytnout přímé měřítko aktivity přímých inhibitorů trombinu.
Test aPTT je široce používán a poskytuje přibližnou indikaci intenzity antikoagulačního účinku dosaženého dabigatranem. Test aPTT se však vyznačuje omezenou citlivostí a není indikován pro přesnou kvantifikaci antikoagulačního účinku, zejména při vysokých plazmatických koncentracích dabigatranem. Zvýšené hodnoty aPTT by měly být interpretovány opatrně.
Obecně lze tvrdit, že tato měření antikoagulační aktivity odrážejí hladiny dabigatranu a mohou poskytnout vodítko pro hodnocení rizika krvácení, tj. Překročení 90. percentilního limitu hladin dabigatranu v době minima nebo naměřeného aPTT. V době minima jsou považovány za související se zvýšeným rizikem krvácení.
Primární prevence epizod žilního tromboembolismu v ortopedické chirurgii
V ustáleném stavu (po 3 dnech) byl geometrický průměr plazmatické koncentrace dabigatranu ve špičce, měřeno přibližně 2 hodiny po podání 220 mg dabigatranetexilátu, 70,8 ng / ml, v rozmezí 35, 2-162 ng / ml ( 25. -75. Percentil).
Geometrický průměr koncentrace dabigatranu v době minima, měřený na konci dávkovacího období (tj. 24 hodin po dávce 220 mg dabigatranu), byl v průměru 22,0 ng / ml, v rozmezí 13, 0-35,7 ng / ml ( 25.-75. Percentil).
U pacientů léčených k prevenci VTE po operaci kyčelního nebo kolenního kloubu 220 mg dabigatran -etexilátu jednou denně,
• 90. percentil plazmatických koncentrací dabigatranu, měřený v době minima (20-28 hodin po předchozí dávce), byl 67 ng / ml (viz body 4.4 a 4.9),
• 90. percentil aPTT v době minima (20–28 hodin po předchozí dávce) byl 51 sekund, neboli 1,3násobek horní hranice normálu.
ECT nebyla měřena u pacientů léčených k prevenci VTE po operaci náhrady kyčle nebo kolena 220 mg dabigatran -etexilátu jednou denně.
Prevence mrtvice a ES u dospělých pacientů s NSAID s jedním nebo více rizikovými faktory
V ustáleném stavu byl geometrický průměr plazmatické koncentrace dabigatranu v době špičky, měřený přibližně 2 hodiny po podání 150 mg dabigatranetexilátu dvakrát denně, 175 ng / ml s rozsahem 117-275 ng / ml (25. -75. Percentil ). Geometrický průměr koncentrace dabigatranu v minimální době, měřený ráno na konci dávkovacího intervalu (tj. 12 hodin po večerní dávce 150 mg dabigatranu), byl v průměru 91,0 ng / ml, s intervalem rovným 61,0- 143 ng / ml (25. -75. Percentil).
U pacientů s NVAF léčených k prevenci mrtvice a SE 150 mg dabigatranetexilátu dvakrát denně,
• 90. percentil plazmatických koncentrací dabigatranu, měřený v době minima (10–16 hodin po předchozí dávce), byl přibližně 200 ng / ml,
• ECT měřená v době minima (10–16 hodin po předchozí dávce), větší než přibližně 3násobek horní hranice normálu, se vztahuje k 90. percentilu pozorovanému při prodloužení ECT o 103 sekund,
• poměr aPTT větší než 2násobek horní hranice normálu (prodloužení aPTT o přibližně 80 sekund) v době minima (10-16 hodin po předchozí dávce) odráží 90. percentil pozorování.
Léčba hluboké žilní trombózy (DVT) a plicní embolie (PE) a prevence recidivy DVT a PE u dospělých (DVT / PE)
U pacientů léčených pro DVT a PE 150 mg dabigatran-etexilátu dvakrát denně, geometrický průměr koncentrace dabigatranu v době minima, měřeno 10-16 hodin po dávce, na konci dávkovacího intervalu (tj. 12 hodin po večeru dávka 150 mg dabigatranu), byla 59,7 ng / ml s rozsahem 38,6 - 94,5 ng / ml (25. -75. percentil) .Pro léčbu DVT a PE dabigatranetexilátem 150 mg dvakrát denně,
• 90. percentil plazmatických koncentrací dabigatranu, měřený v době minima (10–16 hodin po předchozí dávce), byl přibližně 146 ng / ml,
• ECT měřená v době minima (10–16 hodin po předchozí dávce), větší než přibližně 2,3násobek výchozí hodnoty, odkazuje na 90. percentil pozorovaný při prodloužení ECT o 74 sekund,
• 90. percentil aPTT v době minima (10-16 hodin po předchozí dávce) byl 62 sekund, neboli 1,8násobek základní hodnoty.
U pacientů léčených k prevenci recidivy DVT a PE 150 mg dabigatran -etexilátu dvakrát denně nejsou k dispozici žádné farmakokinetické údaje.
Klinická účinnost a bezpečnost
Etnický původ
Mezi kavkazskými, afroamerickými, hispánskými, japonskými nebo čínskými pacienty nebyly pozorovány žádné relevantní etnické rozdíly.
Klinické studie profylaxe venózní tromboembolismu (VTE) po velké chirurgické náhradě kloubu
Ve 2 velkých, randomizovaných, dvojitě zaslepených, dvojitě zaslepených, paralelních studiích s paralelními skupinami byli pacienti naplánovaní na velkou ortopedickou operaci (jedna pro operaci náhrady kolena a jedna pro operaci náhrady kyčelního kloubu) léčeni přípravkem Pradaxa 75 mg nebo 110 mg v rozmezí 1- 4 hodiny po operaci a poté 150 nebo 220 mg jednou denně, hemostáza byla hodnocena normálně nebo 40 mg enoxaparinu den před operací, a proto denně.
Ve studii RE-MODEL (náhrada kolena) byla doba léčby 6–10 dní a ve studii RE-NOVATE (náhrada kyčle) 28–35 dní. Celkem bylo ošetřeno 2076 (náhrada kyčle). Koleno) respektive 3494 (náhrada kyčle) pacientů.
Kombinace všech epizod VTE (včetně PE, proximální a distální DVT, symptomatické i asymptomatické detekovaná rutinní venografií) a mortality ze všech příčin byla primárními cílovými parametry obou studií.
Kombinace všech hlavních epizod VTE (včetně PE, proximální a distální DVT, symptomatické i asymptomatické detekovaná rutinní venografií) a mortality související s VTE byla sekundárním cílovým parametrem považovaným za klinicky významnější.
Výsledky obou studií ukázaly, že antitrombotický účinek přípravku Pradaxa 220 mg a 150 mg nebyl statisticky nižší než u enoxaparinu na celkovou VTE a mortalitu ze všech příčin. Odhadovaný výskyt hlavních epizod VTE je mortalita související s VTE pro Dávka 150 mg byla o něco horší než u enoxaparinu (tabulka 10). Lepší výsledky byly pozorovány u dávky 220 mg, kde byl odhadovaný výskyt hlavních epizod VTE o něco lepší než u enoxaparinu (tabulka 10).
Klinické studie byly provedeny v populaci pacientů s průměrným věkem> 65 let.
V klinických studiích fáze 3 nebyly mezi muži a ženami nalezeny žádné rozdíly v účinnosti a bezpečnosti.
Z populace pacientů účastnících se studií RE-MODEL a RE-NOVATE (5539 léčených pacientů) 51% trpělo souběžnou hypertenzí, 9% souběžným diabetem, 9% onemocněním koronárních tepen a 20% mělo v anamnéze žilní nedostatečnost . Nebyl prokázán žádný z těchto stavů, které by interferovaly s účinky dabigatranu na prevenci VTE nebo frekvenci krvácení.
Údaje pro hlavní cílový parametr VTE a mortality související s VTE byly homogenní s ohledem na primární cílový parametr účinnosti a jsou uvedeny v tabulce 10.
Údaje o koncových bodech pro celkový VTE a úmrtnost všech příčin jsou uvedeny v tabulce 11.
Údaje pro koncové body krvácení, které je považováno za závažné, jsou uvedeny v tabulce 12 níže.
Tabulka 10: Analýza hlavní mortality na VTE a VTE během léčebného období ve studiích ortopedické chirurgie RE-MODEL a RE-NOVATE
Tabulka 11: Analýza celkové VTE a mortality ze všech příčin během léčebného období studií ortopedické chirurgie RE-NOVATE a RE-MODEL
Tabulka 12: Epizody velkého krvácení (ESM) po léčbě v jednotlivých studiích RE-MODEL a RE-NOVATE
Prevence mrtvice a ES u dospělých pacientů s NSAID s jedním nebo více rizikovými faktory
Klinické důkazy o účinnosti dabigatran-etexilátu pocházejí ze studie Randomized Evaluation of Long-term antikoagulant therapy (RE-LY), multicentrické, mezinárodní, randomizované, paralelní skupinové studie porovnávající dvě dávky zaslepeného dabigatran-etexilátu (110 mg a 150 mg dvakrát denně) ve srovnání s otevřeným warfarinem u pacientů s fibrilací síní a středním až vysokým rizikem cévní mozkové příhody a SE. Primárním cílem studie bylo určit, zda dabigatran-etexilát není nižší než warfarin při snižování kompozitního koncového bodu cévní mozkové příhody a SE. Rovněž byla hodnocena statistická nadřazenost.
Ve studii RE-LY bylo randomizováno celkem 18 113 pacientů s průměrným věkem 71,5 roku a průměrným skóre CHADS2 2,1. Populace pacientů byla 64% mužů, 70% bělochů a 16% asiatů. randomizováno na warfarin, průměrné procento času v terapeutickém rozmezí (TTR) (INR 2-3) bylo 64,4% (medián TTR 67%).
Studie RE-LY prokázala, že dabigatranetexilát v dávce 110 mg dvakrát denně není horší než warfarin v prevenci cévní mozkové příhody a SE u subjektů s fibrilací síní, se sníženým rizikem ICH, celkového krvácení a závažného krvácení . Dávka 150 mg dvakrát denně významně snižuje riziko ischemické a hemoragické cévní mozkové příhody, cévní smrti, ICH a celkového krvácení ve srovnání s warfarinem. Výskyt závažného krvácení při této dávce byl srovnatelný s warfarinem. Výskyt infarktu myokardu byl mírně zvýšen při dabigatranetexilátu 110 mg dvakrát denně a 150 mg dvakrát denně ve srovnání s warfarinem (relativní riziko 1,29; p = 0,0929 a relativní riziko 1,27; p = 0,1240) Při lepším monitorování INR byly pozorované přínosy dabigatranetexilátu ve srovnání s poklesem warfarinu.
Tabulky 13-15 ukazují detaily klíčových zjištění v celkové populaci:
Tabulka 13: Analýza výskytu cévní mozkové příhody nebo SE (primární cílový parametr) během období studie RE-LY.
% se vztahuje k ročnímu výskytu události
Tabulka 14: Analýza výskytu ischemické nebo hemoragické mrtvice během období studie RE-LY.
% se vztahuje k ročnímu výskytu události
Tabulka 15: Analýza výskytu úmrtnosti na kardiovaskulární mortalitu ze všech příčin během období studie RE-LY.
% se vztahuje k ročnímu výskytu události
Tabulky 16-18 ukazují primární výsledky účinnosti a bezpečnostní sledovaný parametr u hlavních subpopulací:
Pro primární cílový parametr, cévní mozkovou příhodu a SE nebyly identifikovány žádné podskupiny (tj. Věk, hmotnost, pohlaví, funkce ledvin, etnický původ atd.) S jiným poměrem rizika než warfarin.
Tabulka 16: Relativní riziko a 95% CI mrtvice / SE podle podskupiny
U primárního bezpečnostního parametru velkého krvácení došlo k interakci mezi účinky léčby a věkem. Relativní riziko krvácení se s věkem u dabigatranu zvyšovalo ve srovnání s warfarinem. Relativní riziko bylo vyšší u pacientů ≥ 75 let. Souběžné užívání antiagregačních činidel ASA nebo klopidogrelu přibližně zdvojnásobuje výskyt ESM jak u dabigatranetexilátu, tak u warfarinu. V podskupinách renálních funkcí a skóre CHADS2 nedošlo k žádné významné interakci s účinky léčby.
Tabulka 17: Relativní riziko a 95% CI závažného krvácení podle podskupiny
RELY-ABLE (multicentrická dlouhodobá prodloužená studie léčby dabigatranem u pacientů s fibrilací síní, kteří dokončili studii RE-LY)
Rozšíření studie RE-LY (RELY-ABLE) poskytlo další údaje o bezpečnosti pro kohortu pacientů, kteří pokračovali v užívání stejné dávky dabigatranetexilátu podle přiřazené léčby ve studii RE-LY. Pacienti byli způsobilí. Pro RELY-ABLE pokud v době poslední návštěvy studie RE-LY trvale nepřerušili studijní léčivý přípravek. Zapsaní pacienti nadále dostávali dvojitě zaslepenou dávku stejné dávky dabigatran-etexilátu, při které byli randomizováni v RE- Studie LY, pro prodloužené sledování až 43 měsíců po RE-LY (průměrné sledování RE-LY + RELY-ABLE, 4,5 roku). Bylo zařazeno 5897 pacientů, což představuje 49% pacientů původně randomizovaných pro příjem dabigatranu etexilát ve studii RE-LY a 86% pacientů způsobilých pro RELY-ABLE.
Během dalších 2,5 roku léčby ve studii RELY-ABLE s maximální expozicí delší než 6 let (celková expozice u RE-LY + RELY-ABLE) je dlouhodobý bezpečnostní profil dabigatran-etexilátu potvrzen pro oba 110 mg dvakrát denně a silné dávky 150 mg. Nebyla pozorována žádná nová bezpečnostní data.
Incidence příhod, včetně závažných krvácivých příhod a jiných krvácivých příhod, byla v souladu s incidencí pozorovanou ve studii RE-LY.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Pradaxa ve všech podskupinách pediatrické populace za účelem prevence tromboembolických příhod v povolené indikaci (informace o použití u dětí viz bod 4.2).
Etnický původ (prevence cévní mozkové příhody u pacientů s fibrilací síní)
Mezi kavkazskými, afroamerickými, hispánskými, japonskými nebo čínskými pacienty nebyly pozorovány žádné relevantní etnické rozdíly.
Klinická účinnost a bezpečnost (léčba DVT / PE)
Léčba hluboké žilní trombózy (DVT) a plicní embolie (PE) u dospělých (léčba DVT / PE).
Účinnost a bezpečnost byly studovány ve dvou multicentrických, randomizovaných, dvojitě zaslepených, dvojitých studiích s paralelními skupinami, RE-COVER a RE-COVER II. Tyto studie srovnávaly dabigatranetexilát (150 mg BID) s warfarinem (INR cíl 2, 0-3,0 ) u pacientů s DVT a / nebo akutním PE. Primárním cílem těchto studií bylo zjistit, zda dabigatran-etexilát nebyl při snižování výskytu kompozitního primárního cílového parametru rekurence DVT a / nebo symptomatické PE a ne méně než warfarin úmrtí do 6 měsíců od léčby.
Ve studiích RE-COVER a RE-COVER II bylo randomizováno 5 153 pacientů a bylo léčeno 5 107 pacientů.
Délka léčby fixní dávkou dabigatranu byla 174,0 dnů bez kontroly koagulace. U pacientů randomizovaných na warfarin byl medián času v terapeutickém rozmezí (INR 2,0 až 3,0) 60,6%.
Studie ukázaly, že léčba dabigatranetexilátem 150 mg dvakrát denně nebyla horší než léčba warfarinem (rozpětí non-inferiority pro RE-COVER a RE-COVER II: 3,6 pro rozdíl v riziku a 2,75 podle poměru rizik).
Tabulka 18: Analýza primárních a sekundárních koncových bodů účinnosti (VTE zahrnuje DVT a / nebo PE) až do konce období po léčbě pro studie RE-COVER a RE-COVER II
Etnický původ (léčba DVT / PE)
Mezi kavkazskými, afroamerickými, hispánskými, japonskými nebo čínskými pacienty nebyly pozorovány žádné relevantní etnické rozdíly.
Pediatrická populace (léčba DVT / PE)
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Pradaxa u všech podskupin pediatrické populace v léčbě DVT / PE (informace o použití u dětí viz bod 4.2).
Farmakokinetika a farmakodynamika dabigatran-etexilátu podávaného dvakrát denně po tři po sobě jdoucí dny (celkem 6 dávek) na konci standardní antikoagulační léčby byla hodnocena v otevřené studii bezpečnosti a snášenlivosti provedené u 9 klinicky stabilních dospívajících (věk 12 až
Klinická účinnost a bezpečnost (prevence DVT / PE)
Prevence recidivy hluboké žilní trombózy (DVT) a plicní embolie (PE) u dospělých (prevence DVT / PE)
Byly provedeny dvě randomizované, dvojitě zaslepené studie s paralelními skupinami u pacientů dříve léčených antikoagulační léčbou. Warfarinem kontrolovaná studie RE-MEDY zahrnovala pacienty již léčené 3 až 12 měsíců a vyžadující další antikoagulační léčbu, placebem kontrolovaná studie RE-SONATE zahrnovala pacienty již léčené 6 až 18 měsíců inhibitory vitaminu K
Cílem studie RE-MEDY bylo porovnat bezpečnost a účinnost perorálního dabigatranetexilátu (150 mg dvakrát denně) s warfarinem (cílová hodnota INR 2,0-3,0) pro dlouhodobou léčbu a prevenci relapsů symptomatické DVT a / nebo PE. Bylo randomizováno celkem 2 866 pacientů a léčeno 2 856 pacientů. Délka léčby dabigatranetexilátem se pohybovala od 6 do 36 měsíců (medián 534,0 dnů). U pacientů randomizovaných na warfarin byl medián času v terapeutickém rozmezí (INR 2,0-3,0) 64,9%.
Studie RE-MEDY prokázala, že léčba dabigatranetexilátem 150 mg dvakrát denně nebyla horší než warfarin (rozpětí noninferiority: 2,85 pro poměr rizik a 2,8 pro rozdíl rizik).
Tabulka 19: Analýza primárních a sekundárních cílových parametrů účinnosti (VTE zahrnuje DVT a / nebo PE) až do konce období po léčbě pro studii RE-MEDY
Cílem studie RE-SONATE bylo vyhodnotit nadřazenost dabigatranetexilátu oproti placebu v prevenci relapsu symptomatické DVT a / nebo PE u pacientů, kteří již absolvovali 6 až 8 měsíců léčby AVK. Terapie zahrnovala 6 měsíců. s dabigatranetexilátem 150 mg dvakrát denně bez nutnosti sledování.
Studie RE-SONATE prokázala, že dabigatran-etexilát byl v prevenci recidivy symptomatických epizod DVT / PE včetně úmrtí z nevysvětlených příčin lepší než placebo, se snížením rizika z 5,6% na 0,4% (relativní snížení 92% rizika na základě poměru rizika) během doba léčby (str
Studie zahrnovala observační období sledování 12 měsíců po ukončení léčby. Po přerušení studie byl účinek léčivého přípravku zachován až do konce období sledování, což naznačuje, že počáteční účinek léčby dabigatranem přetrvává. Nebyl pozorován žádný rebound efekt. Na konci období sledování byly příhody VTE u pacientů léčených dabigatranetexilátem 6,9% vs 10,7% ve srovnání se skupinou s placebem (poměr rizik 0,61 (95% CI 0,42,0, 88), p = 0,0082).
Tabulka 20: Analýza primárních a sekundárních koncových bodů účinnosti (VTE zahrnuje DVT a / nebo PE) až do konce období po léčbě pro studii RE-SONATE
Etnický původ (prevence DVT / PE)
Nebyly pozorovány žádné klinicky relevantní etnické rozdíly mezi kavkazskými, afroamerickými, hispánskými, japonskými nebo čínskými pacienty.
Pediatrická populace (prevence DVT / PE)
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Pradaxa ve všech podskupinách pediatrické populace v prevenci DVT / PE (informace o použití u dětí viz bod 4.2).
Klinické studie prevence tromboembolie u pacientů s protézami srdečních chlopní
Studie fáze II hodnotila dabigatran -etexilát a warfarin u celkem 252 pacientů, kteří podstoupili částečně nedávnou chirurgickou operaci chlopně (tj. Zapsaní během hospitalizace) a částečně mechanickou operaci srdeční chlopně déle než tři měsíce. U dabigatran -etexilátu bylo pozorováno více tromboembolických příhod (hlavně cévní mozkové příhody a symptomatické / asymptomatické trombózy chlopní) a více krvácivých příhod než u warfarinu. U bezprostředně pooperačních pacientů se velké krvácení projevovalo hlavně jako hemoragické perikardiální výpotky, zejména u pacientů, kteří začali s dabigatranetexilátem krátce (tj. 3. den) po operaci protézy srdeční chlopně (viz bod 4.3).
05.2 Farmakokinetické vlastnosti
Po perorálním podání je dabigatran -etexilát rychle a úplně přeměněn na dabigatran, což je aktivní forma v plazmě. Převažující metabolickou reakcí je štěpení prekurzoru dabigatranetexilátu hydrolýzou katalyzovanou esterázou na účinnou látku dabigatran. Absolutní biologická dostupnost dabigatranu po perorálním podání přípravku Pradaxa je přibližně 6,5%.
Po perorálním podání přípravku Pradaxa zdravým dobrovolníkům je farmakokinetický profil dabigatranu v plazmě charakterizován rychlým zvýšením plazmatických koncentrací s Cmax dosaženým 0,5 - 2,0 hodiny po podání.
Vstřebávání
Studie hodnotící pooperační absorpci dabigatran-etexilátu, 1-3 hodiny po operaci, prokázala relativně pomalou absorpci ve srovnání se zdravými dobrovolníky, což dokazuje profil plazmatické koncentrace v čase bez vysokých maximálních plazmatických koncentrací. Maximálních plazmatických koncentrací je dosaženo 6 hodin po podání v pooperačním období v důsledku faktorů, jako je anestezie, střevní paréza a chirurgické účinky, bez ohledu na orální formulaci léčivého přípravku. V další studii bylo ukázáno, že pomalá a opožděná absorpce se obvykle vyskytuje pouze v den operace.V následujících dnech je absorpce dabigatranu rychlá s maximální plazmatickou koncentrací dosaženou 2 hodiny po podání léčiva.
Jídlo nemění biologickou dostupnost dabigatran -etexilátu, ale prodlužuje čas dosažení maximální plazmatické koncentrace o 2 hodiny.
Pokud jsou pelety odebrány bez kapsle HPMC (hydroxypropylmethylcelulóza), může se orální biologická dostupnost zvýšit o 75% ve srovnání s referenční formulací s kapslí. Během klinického použití proto musí být vždy zachována celistvost tobolek HPMC, aby se zabránilo neúmyslnému zvýšení biologické dostupnosti dabigatran -etexilátu. Pacienti by proto měli být poučeni, aby neotvírali tobolky a nebrali jejich obsah samotný (např. Sypali do jídla nebo nalili do nápoje) (viz bod 4.2).
Rozdělení
Byla pozorována nezávislá vazba dabigatranu na proteiny lidské plazmy na nízké koncentraci (34-35%). Distribuční objem dabigatranu 60 - 70 l převyšuje objem celkových tělesných tekutin, což ukazuje na mírnou distribuci dabigatranu do tkání.
Cmax a plocha pod křivkou plazmatické koncentrace v čase byly úměrné dávce.Plazmatické koncentrace dabigatranu vykazovaly bi-exponenciální pokles s průměrným terminálním poločasem 11 hodin u zdravých starších osob. Po více dávkách byl pozorován terminální poločas přibližně 12 až 14 hodin. Poločas byl nezávislý na dávce. Poločas se prodlužuje, pokud je poškozena funkce ledvin, jak ukazuje tabulka 21.
Biotransformace
Metabolismus a vylučování dabigatranu byly studovány po podání jedné intravenózní dávky radioaktivního dabigatranu zdravým mužským subjektům. Po intravenózní dávce byla radioaktivita dabigatranu eliminována primárně močí (85%). Vylučování stolicí bylo odhadnuto na 6% podané dávky. Celkové zotavení radioaktivity se pohybovalo od 88 do 94% podané dávky do 168 hodin po podání.
Dabigatran podléhá konjugaci za vzniku farmakologicky aktivních acylglukuronidů. Existují čtyři poziční izomery 1-O, 2-O, 3-O, 4-O acylglukuronidů, z nichž každý je odhadován na méně než 10% celkového dabigatranu v plazmě. Stopy dalších metabolitů jsou detekovatelné pouze vysoce citlivými analytickými metodami. Dabigatran je vylučován převážně beze změny močí rychlostí přibližně 100 ml / min, což odpovídá rychlosti glomerulární filtrace.
Zvláštní populace
Selhání ledvin
Ve studiích fáze I je expozice (AUC) dabigatranu po perorálním podání přípravku Pradaxa přibližně 2,7krát vyšší u dobrovolníků se středně těžkou poruchou funkce ledvin (CrCL mezi 30 a 50 ml / min) než u dobrovolníků bez poruchy funkce ledvin.
U malého počtu dobrovolníků s těžkou renální insuficiencí (CrCL 10 - 30 ml / min) byla expozice dabigatranu (AUC) přibližně 6krát vyšší a poločas přibližně 2krát delší, než byl pozorován v populaci bez renální insuficience. (Viz. oddíly 4.2, 4.3 a 4.4).
Tabulka 21: Poločas celkového dabigatranu u zdravých subjektů a subjektů s poruchou funkce ledvin.
Clearance dabigatranu hemodialýzou byla zkoumána u 7 pacientů s terminálním chronickým selháním ledvin (ESRD) bez fibrilace síní. Dialýza byla prováděna průtokem dialyzátu 700 ml / min po dobu čtyř hodin a průtokem krve jak 200 ml / min, tak 350-390 ml / min. Výsledkem bylo odstranění 50% až 60% koncentrací dabigatranu. Množství látky odebrané dialýzou je úměrné průtoku krve až 300 ml / min. Antikoagulační aktivita dabigatranu klesala se snižujícími se plazmatickými koncentracemi a farmakokinetický / farmakodynamický vztah nebyl postupem změněn.
Medián CrCL ve studii RE-LY byl 68,4 ml / min. Téměř polovina (45,8%) pacientů s RE -LY měla CRL> 50 -
Medián CrCL ve studii RE-COVER byl 100,4 ml / min. 21,7% pacientů mělo mírné poškození ledvin (CrCL> 50 - 80 ml / min. Podobné hodnoty CrCL byly zjištěny ve studii RE -COVER II.
Medián CrCL ve studiích RE-MEDY a RE-SONATE byl 99,0 ml / min, respektive 99,7 ml / min. 22,9% a 22,5% pacientů mělo CRL> 50-
Starší pacienti
Specifické farmakokinetické studie fáze I provedené u starších subjektů prokázaly 40 až 60% nárůst AUC a více než 25% Cmax ve srovnání s mladými subjekty.
Vliv věku na expozici dabigatranu byl potvrzen ve studii RE-LY s vyšší minimální koncentrací přibližně 31% u subjektů ve věku ≥ 75 let a s nižší minimální koncentrací přibližně 22% u subjektů ve věku
Porucha funkce jater
Nebyla zjištěna žádná změna expozice dabigatranu u 12 subjektů se středně těžkou poruchou funkce jater (Child Pugh B) ve srovnání s 12 kontrolními subjekty (viz body 4.2 a 4.4).
Tělesná hmotnost
Koncentrace dabigatranu v době minima byly přibližně o 20% nižší u pacientů s tělesnou hmotností> 100 kg ve srovnání s pacienty s tělesnou hmotností mezi 50 a 100 kg. Většina pacientů (80,8%) měla tělesnou hmotnost ≥ 50 kg a
Typ
Expozice léčivé látce v primární prevenci epizod VTE byla přibližně o 40% až 50% vyšší u pacientek a nedoporučuje se úprava dávkování. U pacientek s fibrilací síní byla expozice účinné látce o 30% vyšší než v pacientů mužského pohlaví jak v době minima, tak po podání dávky. Nedoporučuje se žádná úprava dávky (viz bod 4.2).
etnické pozadí
Pokud jde o farmakokinetiku a farmakodynamiku dabigatranu, nebyly mezi kavkazskými, afroamerickými, hispánskými, japonskými nebo čínskými pacienty pozorovány žádné relevantní mezietnické rozdíly.
Farmakokinetické interakce
Proléčivý dabigatran-etexilát je substrátem efluxního transportéru P-gp, nikoli však dabigatranu. Z tohoto důvodu současné použití s inhibitory transportéru P-gp (amiodaron, verapamil, klarithromycin, chinidin, donedaron, tikagrelor a ketokonazol) a s induktory (rifampicin) (viz body 4.2, 4.4 a 4.5).
Interakční studie in vitro nevykazovaly žádnou inhibici ani indukci hlavních izoenzymů cytochromu P450. To potvrdily studie in vivo provedené na zdravých dobrovolnících, ve kterých nebyla prokázána žádná interakce mezi touto léčbou a následujícími účinnými látkami: atorvastatin (CYP3A4), digoxin (interakce s transportérem P-gp) a diklofenak (CYP2C9).
05.3 Předklinické údaje vztahující se k bezpečnosti
Údaje z neklinických studií na základě konvenčních studií farmakologické bezpečnosti, toxicity po opakovaném podávání a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Účinky pozorované ve studiích toxicity po opakovaném podávání byly způsobeny zesíleným farmakodynamickým účinkem dabigatranu.
Účinek na ženskou plodnost ve formě snížené implantace a zvýšené ztráty před implantací byl pozorován při dávkách 70 mg / kg (5násobek hladiny plazmatické expozice u pacientů). Při dávkách toxických pro matku (5 až 10násobek plazmatické expozice u pacientů) byl u potkanů a králíků pozorován pokles tělesné hmotnosti a životaschopnosti plodu se zvýšením změn plodu. V prenatální a postnatální studii bylo pozorováno zvýšení úmrtnosti plodu při dávkách toxických pro matku (dávka odpovídající hladině plazmatické expozice 4krát vyšší než u pacientů).
Ve studiích celoživotní toxicity na potkanech a myších nebyl prokázán tumorigenní potenciál dabigatranu až do maximální dávky 200 mg / kg.
Dabigatran, aktivní molekula dabigatran -etexilát mesylátu, je v životním prostředí perzistentní.
06.0 FARMACEUTICKÉ INFORMACE
06.1 Pomocné látky
Obsah tobolky
• Kyselina vinná
• arabská guma
• Hypromelóza
• Dimethicone 350
• Mastek
• Hydroxypropylcelulóza
Kapsle
• Karagenan
• Chlorid draselný
• Oxid titaničitý
• Indigokarmín (E132)
• Žlutá při západu slunce (E110)
• Hypromelóza
Černý inkoust pro tisk
• Šelak
• Černý oxid železitý (E172)
• Hydroxid draselný
06.2 Neslučitelnost
Irelevantní.
06.3 Doba platnosti
Blistr a lahvička: 3 roky.
Jakmile je lahvička otevřena, měl by být léčivý přípravek spotřebován do 4 měsíců.
06.4 Zvláštní opatření pro skladování
Blistr
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Láhev
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
06.5 Charakter vnitřního obalu a obsah balení
Balení obsahující 10 x 1, 30 x 1 nebo 60 x 1 tvrdou tobolku, vícečetné balení obsahující 3 balení po 60 x 1 tvrdých tobolek (180 tvrdých tobolek) a vícečetné balení obsahující 2 balení po 50 x 1 tvrdých tobolek (100 tvrdých tobolek)) . Kromě toho balení obsahující 6 bílých hliníkových blistrů, dělitelných na jednotlivou dávku (60 x 1). Blistr se skládá z horní vrstvy hliníku potažené kopolymery polyvinylchlorid-polyvinylacetát (akryláty PVCAC) ve styku s přípravkem a spodní vrstvy hliníku potažené polyvinylchloridem (PVC) ve styku s přípravkem.
Polypropylenová láhev se šroubovacím uzávěrem obsahující 60 tvrdých tobolek.
Na trhu nemusí být všechny velikosti balení.
06.6 Návod k použití a zacházení
Při používání přípravku Pradaxa baleného v blistrech je třeba dodržovat následující pokyny:
• Tvrdá tobolka musí být vyjmuta z blistru zvednutím hliníkové fólie na zadní straně.
• Tvrdá tobolka nesmí být protlačována přes blistr.
• Hliníkovou fólii blistru je třeba zvedat pouze v případě, že je zapotřebí tvrdá tobolka.
Při používání kapslí balených v lahvích je třeba dodržovat následující pokyny:
• Láhev se otevírá stisknutím a otočením víčka.
Nepoužitý léčivý přípravek a odpad z tohoto přípravku musí být zlikvidován v souladu s místními předpisy.
07.0 DRŽITEL ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH
Binger Str. 173
D-55216 Ingelheim am Rhein
Německo
08.0 REGISTRAČNÍ ČÍSLO
EU/1/08/442/005
038451050
EU/1/08/442/006
038451062
EU/1/08/442/007
038451074
EU/1/08/442/008
038451086
EU/1/08/442/014
038451148
EU/1/08/442/015
EU/1/08/442/018
09.0 DATUM PRVNÍ REGISTRACE NEBO PRODLOUŽENÍ REGISTRACE
Datum první registrace: 18. března 2008
Datum posledního obnovení: 17. ledna 2013
10.0 DATUM REVIZE TEXTU
18. prosince 2014







-e-sali.jpg)