Aktivní složky: Sitagliptin
Januvia 25 mg potahované tablety
Příbalové letáky Januvia jsou k dispozici pro velikosti balení:- Januvia 25 mg potahované tablety
- Januvia 50 mg potahované tablety
- Januvia 100 mg potahované tablety
Proč se přípravek Januvia používá? K čemu to je?
Přípravek Januvia obsahuje léčivou látku sitagliptin, která patří do třídy léků nazývaných inhibitory dipeptidyl peptidázy-4 (DPP-4), které snižují hladinu cukru v krvi u dospělých pacientů s diabetes mellitus 2. typu.
Tento léčivý přípravek pomáhá zvyšovat hladiny inzulinu produkovaného po jídle a snižuje množství cukru produkovaného tělem.
Váš lékař vám předepsal tento lék, aby vám pomohl snížit hladinu cukru v krvi, která je příliš vysoká v důsledku cukrovky typu 2. Tento lék lze použít samostatně nebo společně s jinými léky (inzulín, metformin, sulfonylmočovina nebo glitazony), které snižují hladinu cukru v krvi, může již užívat k léčbě cukrovky spolu s dietou a cvičebním programem.
Co je diabetes typu 2?
Cukrovka 2. typu je onemocnění, při kterém tělo nevytváří dostatek inzulinu a inzulín produkovaný tělem nepracuje tak, jak by měl. Tělo si také dokáže vyrobit příliš mnoho cukru. Když k tomu dojde, v krvi se hromadí cukr (glukóza). To může vést k vážným zdravotním problémům, jako jsou srdeční choroby, onemocnění ledvin, slepota a amputace.
Kontraindikace Kdy by Januvia neměla být použita
Neužívejte přípravek Januvia
- jestliže jste alergický (á) na sitagliptin nebo na kteroukoli další složku tohoto přípravku
Opatření pro použití Co potřebujete vědět před užitím přípravku Januvia
U pacientů léčených přípravkem Januvia byly hlášeny případy zánětu slinivky břišní (pankreatitida) (viz bod 4).
Informujte svého lékaře, pokud máte nebo jste měl:
- onemocnění slinivky břišní (jako je pankreatitida)
- žlučové kameny, závislost na alkoholu nebo velmi vysoké hladiny triglyceridů (forma tuku) v krvi. Tyto zdravotní stavy mohou zvýšit riziko vzniku pankreatitidy (viz bod 4).
- diabetes 1. typu
- diabetická ketoacidóza (komplikace diabetu s vysokou hladinou cukru v krvi, rychlým úbytkem hmotnosti, nevolností nebo zvracením)
- jakékoli minulé nebo současné problémy s ledvinami
- alergická reakce na přípravek Januvia (viz bod 4).
Je nepravděpodobné, že by tento lék způsoboval nízkou hladinu cukru v krvi (hypoglykémii), protože při nízké hladině cukru v krvi nefunguje. Pokud je však tento lék podáván se sulfonylureou nebo s inzulínem, může dojít k hypoglykémii. Váš lékař může snížit dávku sulfonylurey nebo inzulínu.
Děti a dospívající
Děti a mladiství do 18 let by tento přípravek neměli používat. Není známo, zda je použití tohoto léku bezpečné a účinné u dětí a dospívajících do 18 let.
Interakce Které léky nebo potraviny mohou změnit účinek přípravku Januvia
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval (a) nebo které možná budete užívat.
Zejména informujte svého lékaře, pokud užíváte digoxin (lék používaný k léčbě nepravidelného srdečního tepu a dalších srdečních problémů). Pokud užíváte přípravek Januvia, může být nutné zkontrolovat hladinu digoxinu v krvi.
Varování Je důležité vědět, že:
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat. Tento přípravek byste neměli užívat během těhotenství.
Není známo, zda tento léčivý přípravek přechází do mateřského mléka. Tento lék byste neměli užívat, pokud kojíte nebo si myslíte, že kojit budete muset.
Řízení dopravních prostředků a obsluha strojů
Tento léčivý přípravek nemá žádný nebo má zanedbatelný vliv na schopnost řídit a obsluhovat stroje. Byly však hlášeny závratě a somnolence, které mohou ovlivnit vaši schopnost řídit a obsluhovat stroje.
Užívání tohoto léku s jinými léky nazývanými deriváty sulfonylurey nebo s inzulínem může způsobit hypoglykémii, která může ovlivnit vaši schopnost řídit, obsluhovat stroje nebo pracovat bez ochranných bariér.
Dávka, způsob a doba podání Jak se přípravek Januvia používá: Dávkování
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. V případě pochybností se poraďte se svým lékařem nebo lékárníkem.
Obvyklá doporučená dávka je:
- jedna 100 mg potahovaná tableta
- jednou denně
- pusou
Pokud máte problémy s ledvinami, lékař vám může předepsat nižší dávky (například 25 mg nebo 50 mg).
Tento lék můžete užívat s jídlem a pitím nebo bez nich.
Váš lékař vám může tento lék předepsat samostatně nebo společně s jinými léky, které snižují hladinu cukru v krvi.
Dieta a cvičení mohou vašemu tělu pomoci lépe využívat krevní cukr. Během užívání přípravku Januvia je důležité dodržovat dietu a cvičební program doporučený lékařem.
Předávkování Co dělat, když jste užil příliš mnoho přípravku Januvia
Jestliže jste užil více přípravku Januvia, než jste měl
Pokud užijete více než předepsanou dávku tohoto léku, okamžitě kontaktujte svého lékaře.
Jestliže jste zapomněl (a) užít přípravek Januvia
Pokud zapomenete dávku, vezměte si ji, jakmile si vzpomenete. Pokud si nevzpomenete, dokud nedojde k další dávce, vynechejte zapomenutou dávku a pokračujte v normální dávce. Nezdvojnásobujte dávku tohoto léku.
Jestliže jste přestal (a) užívat přípravek Januvia
Pokračujte v užívání tohoto léku tak dlouho, jak vám to lékař předepisuje, abyste mohli i nadále sledovat hladinu cukru v krvi. Tento lék byste neměli přestat užívat, aniž byste si nejprve promluvili se svým lékařem.
Máte -li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Nežádoucí účinky Jaké jsou vedlejší účinky přípravku Januvia
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
PŘESTAŇTE užívat přípravek Januvia a okamžitě kontaktujte lékaře, pokud zaznamenáte některý z následujících závažných nežádoucích účinků:
- Silná a přetrvávající bolest v břiše (oblast žaludku), která se může rozšířit do zad s nevolností a zvracením nebo bez nich, protože to mohou být příznaky zánětu slinivky břišní (pankreatitida).
Pokud máte závažnou alergickou reakci (frekvence není známa), včetně vyrážky, kopřivky, puchýřů na kůži / odlupující se kůže a otoku obličeje, rtů, jazyka a hrdla, které mohou způsobit potíže s dýcháním nebo polykáním, ukončete léčbu tímto přípravkem a okamžitě kontaktujte svého lékaře. Lékař vám může předepsat lék k léčbě alergické reakce a jiný lék na cukrovku. U některých pacientů se po přidání sitagliptinu k metforminu vyskytly následující nežádoucí účinky:
Časté (mohou postihnout až 1 z 10 lidí): nízká hladina cukru v krvi, nevolnost, plynatost, zvracení.
Méně časté (mohou postihnout až 1 ze 100 lidí): bolest žaludku, průjem, zácpa, ospalost.
Někteří pacienti hlásili různé typy bolesti žaludku při zahájení léčby sitagliptinem a metforminem společně v rámci kombinované terapie (frekvence je běžná).
U některých pacientů se při užívání sitagliptinu v kombinaci se sulfonylureou a metforminem vyskytly následující nežádoucí účinky:
Velmi časté (mohou postihnout více než 1 z 10 lidí): nízká hladina cukru v krvi.
Časté: zácpa.
U některých pacientů se při užívání sitagliptinu a pioglitazonu vyskytly následující nežádoucí účinky:
Časté: plynatost, otoky rukou nebo nohou.
U některých pacientů se při užívání sitagliptinu v kombinaci s pioglitazonem a metforminem vyskytly následující nežádoucí účinky:
omune: otok rukou nebo nohou.
U některých pacientů se při užívání sitagliptinu v kombinaci s inzulínem (s nebo bez metforminu) vyskytly následující nežádoucí účinky:
Časté: chřipka.
Méně časté: sucho v ústech.
Někteří pacienti zaznamenali následující nežádoucí účinky při užívání samotného sitagliptinu v klinických studiích nebo při samotném použití po schválení a / nebo s jinými léky na diabetes:
Časté: nízká hladina cukru v krvi, bolest hlavy, infekce horních cest dýchacích, rýma nebo ucpaný nos a bolest v krku, artróza, bolest paží nebo nohou.
Méně časté: závratě, zácpa.
Frekvence není známa: problémy s ledvinami (někdy vyžadující dialýzu); Zvracel; bolest kloubů; svalová bolest; bolest zad; intersticiální plicní nemoc.
Hlášení nežádoucích účinků
Pokud se u vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Nežádoucí účinky můžete hlásit také přímo na adresu: Státní ústav pro kontrolu léčiv Šrobárova 48100 41 Praha 10 Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
Expirace a retence
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na blistru a krabičce za „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání
Nevyhazujte žádné léky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Pomůže to chránit životní prostředí.
Co přípravek Januvia obsahuje
- Léčivou látkou je sitagliptin. Jedna potahovaná tableta (tableta) obsahuje monohydrát sitagliptin-fosfátu, což odpovídá 25 mg sitagliptinu.
- Dalšími složkami jsou: v jádru tablety: mikrokrystalická celulóza (E460), bezvodý hydrogenfosforečnan vápenatý (E341), sodná sůl kroskarmelózy (E468), stearát hořečnatý (E470b) a stearylfumarát sodný. Potah tablety obsahuje: polyvinylalkohol, makrogol 3350, mastek (E553b), oxid titaničitý (E171), červený oxid železitý (E172) a žlutý oxid železitý (E172).
Jak přípravek Januvia vypadá a co obsahuje toto balení
Kulaté, růžové potahované tablety s „221“ na jedné straně.
Neprůhledné blistry (PVC / PE / PVDC a hliník). Balení 14, 28, 30, 56, 84, 90 nebo 98 potahovaných tablet a 50 x 1 potahovaných tablet v perforovaných jednodávkových blistrech.
Na trhu nemusí být všechny velikosti balení.
Zdroj příbalové informace: AIFA (Italská agentura pro léčivé přípravky). Obsah zveřejněný v lednu 2016. Přítomné informace nemusí být aktuální.
Chcete-li mít přístup k nejaktuálnější verzi, doporučujeme navštívit webovou stránku AIFA (Italská agentura pro léčivé přípravky). Prohlášení a užitečné informace.
01.0 NÁZEV LÉČIVÉHO PŘÍPRAVKU
TABLETY JANUVIA 25 MG potažené filmem
02.0 KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tableta obsahuje monohydrát sitagliptin -fosfátu, což odpovídá sitagliptinum 25 mg.
Úplný seznam pomocných látek viz bod 6.1.
03.0 LÉKOVÁ FORMA
Potahovaná tableta (tableta).
Kulaté, růžové potahované tablety s „221“ na jedné straně.
04.0 KLINICKÉ INFORMACE
04.1 Terapeutické indikace
U dospělých pacientů s diabetes mellitus 2. typu je přípravek Januvia indikován ke zlepšení kontroly glykémie:
v monoterapii
• u pacientů nedostatečně kontrolovaných pouze dietou a cvičením a pro něž metformin není vhodný z důvodu kontraindikací nebo nesnášenlivosti.
v duální orální terapii v kombinaci s
• metformin, pokud dieta a cvičení plus samotný metformin neposkytují dostatečnou kontrolu glykémie.
• derivát sulfonylurey, pokud dieta a cvičení plus maximální tolerovaná dávka sulfonylurey samotné neposkytují adekvátní kontrolu glykémie a pokud metformin není vhodný z důvodu kontraindikací nebo intolerance.
• agonista receptoru gama (PPARγ) aktivovaného proliferátorem peroxizomů (např. Thiazolidindion), pokud je vhodné použití agonisty PPARγ a pokud dieta a cvičení plus samotný agonista PPARγ neposkytují adekvátní kontrolní hladinu cukru v krvi.
v trojité orální terapii v kombinaci s
• sulfonylmočovina a metformin, pokud dieta a cvičení plus duální terapie těmito léky neposkytují adekvátní kontrolu glykémie.
• agonista PPARγ a metformin, pokud je použití agonisty PPARγ vhodné a pokud dieta a cvičení plus duální terapie těmito léčivými přípravky neposkytují adekvátní kontrolu glykémie.
Přípravek Januvia je také indikován jako doplňková terapie k inzulinu (s metforminem nebo bez metforminu), pokud dieta a cvičení plus stabilní dávka inzulinu neposkytují adekvátní kontrolu glykémie.
04.2 Dávkování a způsob podání
Dávkování
Dávka je 100 mg sitagliptinu jednou denně. Při použití v kombinaci s metforminem a / nebo agonistou PPARγ by měla být dávka metforminu a / nebo agonisty PPARγ zachována a přípravek Januvia by měl být podáván souběžně.
Pokud se přípravek Januvia používá v kombinaci se sulfonylmočovinou nebo inzulínem, lze ke snížení rizika hypoglykémie zvážit nižší dávku sulfonylurey nebo inzulinu (viz bod 4.4).
Pokud vynecháte dávku přípravku Januvia, měla by být podána, jakmile si pacient vzpomene.
Ve stejný den by neměla být podána dvojnásobná dávka.
Zvláštní populace
Poškození ledvin
Při zvažování použití sitagliptinu v kombinaci s jiným antidiabetickým léčivým přípravkem by měl být zkontrolován způsob použití u pacientů s poruchou funkce ledvin.
U pacientů s mírnou poruchou funkce ledvin (clearance kreatininu [CrCl] ≥ 50 ml / min) není úprava dávky nutná.
U pacientů se středně těžkou poruchou funkce ledvin (CrCl ≥ 30 až
U pacientů s těžkou poruchou funkce ledvin (CrCl hemodialýza nebo peritoneální dialýza je dávka přípravku Januvia 25 mg jednou denně. Léčbu lze podávat bez ohledu na načasování dialýzy.
Protože dochází k úpravě dávkování na základě funkce ledvin, doporučuje se před zahájením léčby přípravkem Januvia a poté pravidelně pravidelně hodnotit renální funkce.
Porucha funkce jater
U pacientů s mírnou až středně těžkou poruchou funkce jater není nutná úprava dávky. Přípravek Januvia nebyl studován u pacientů s těžkou poruchou funkce jater a je nutná opatrnost (viz bod 5.2).
Jelikož je však sitagliptin eliminován primárně ledvinami, neočekává se, že by závažné poškození jater ovlivnilo farmakokinetiku sitagliptinu.
Senioři
Na základě věku není nutná žádná úprava dávky.U pacientů ve věku ≥ 75 let jsou k dispozici omezené údaje o bezpečnosti a v těchto případech je nutná opatrnost.
Pediatrická populace
Bezpečnost a účinnost sitagliptinu u dětí a dospívajících mladších 18 let Nejsou k dispozici žádné údaje.
Způsob podání
Přípravek Januvia lze užívat s jídlem nebo bez jídla.
04.3 Kontraindikace
Přecitlivělost na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 (viz body 4.4 a 4.8).
04.4 Zvláštní upozornění a vhodná opatření pro použití
Všeobecnost
Přípravek Januvia by neměli používat pacienti s diabetem 1. typu ani k léčbě diabetické ketoacidózy.
Akutní pankreatitida
Použití inhibitorů dipeptyl-peptidázy 4 (DPP-4) je spojeno s rizikem vzniku akutní pankreatitidy.Pacienti by měli být informováni o charakteristickém příznaku akutní pankreatitidy: závažné, trvalé bolesti břicha. Úzkost pankreatitidy byla pozorována po přerušení léčby sitagliptinem (s podpůrnou léčbou nebo bez ní), ale byly hlášeny velmi vzácné případy nekrotizující nebo hemoragické pankreatitidy a / nebo úmrtí. Pokud je podezření na pankreatitidu, léčba přípravkem Januvia a dalšími potenciálně podezřelými léčivými přípravky by měla být přerušena; pokud se potvrdí diagnóza akutní pankreatitidy, léčba přípravkem Januvia by neměla být znovu zahájena. Opatrnosti je třeba u pacientů s anamnézou pankreatitidy.
Hypoglykémie při použití v kombinaci s jinými antihyperglykemickými léky
V klinických studiích přípravku Januvia v monoterapii a jako součást kombinované terapie s léčivými přípravky, o nichž není známo, že by způsobovaly hypoglykémii (např. Metformin a / nebo agonista PPARγ), byl výskyt hypoglykémie hlášený u sitagliptinu podobný výskytu u pacientů užívajících placebo. Při použití sitagliptinu v kombinaci s inzulínem nebo derivátem sulfonylurey byla pozorována hypoglykémie. Proto lze ke snížení rizika hypoglykémie zvážit nižší dávku sulfonylurey nebo inzulinu (viz bod 4.2).
Poškození ledvin
Sitagliptin se vylučuje ledvinami. Aby se dosáhlo plazmatických koncentrací sitagliptinu podobných těm u pacientů s normální funkcí ledvin, doporučují se nižší dávky u pacientů se středně těžkou a těžkou poruchou funkce ledvin, stejně jako u pacientů s ESRD vyžadujícím hemodialýzu nebo peritoneální dialýzu (viz body 4.2 a 5.2).
Při zvažování použití sitagliptinu v kombinaci s jiným antidiabetickým léčivým přípravkem by měl být zkontrolován způsob použití u pacientů s poruchou funkce ledvin.
Reakce přecitlivělosti
Ve zprávách po uvedení přípravku na trh byly u pacientů léčených sitagliptinem hlášeny závažné reakce z přecitlivělosti. Tyto reakce zahrnují anafylaxi, angioedém a exfoliativní kožní poruchy včetně Stevens-Johnsonova syndromu. Nástup těchto reakcí nastal během prvních 3 měsíců po zahájení léčby, přičemž některé zprávy se objevily po první dávce.
Pokud je podezření na hypersenzitivní reakci, léčba přípravkem Januvia by měla být ukončena. Je třeba prozkoumat další možné příčiny události a zahájit alternativní léčbu diabetu.
04.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Účinky jiných léčivých přípravků na sitagliptin
Níže popsané klinické údaje naznačují, že riziko klinicky významných interakcí se souběžně podávanými léčivými přípravky je omezené.
Vzdělávání in vitro naznačil, že hlavním enzymem odpovědným za omezený metabolismus sitagliptinu je CYP3A4 s přispěním CYP2C8. U pacientů s normální funkcí ledvin má metabolismus, včetně metabolismu CYP3A4, omezenou roli při clearance sitagliptinu. Metabolismus může hrát významnější roli při eliminaci sitagliptinu v kontextu závažného poškození ledvin nebo konečného stadia onemocnění ledvin (ESRD) .Z tohoto důvodu je možné, že silné inhibitory CYP3A4 (např. Ketokonazol, itrakonazol, ritonavir, klarithromycin) mohou změnit farmakokinetika sitagliptinu u pacientů s těžkou poruchou funkce ledvin nebo ESRD Účinky silných inhibitorů CYP3A4 na poškození ledvin nebyly v klinické studii stanoveny.
Dopravní studie in vitro ukázal, že sitagliptin je substrátem pro p-glykoprotein e
pro transportér organických aniontů 3 (OAT3). Transport sitagliptinu zprostředkovaný OAT3 byl inhibován in vitro probenecid, ačkoli riziko klinicky relevantních interakcí je považováno za omezené. Souběžné podávání inhibitorů OAT3 nebylo hodnoceno in vivo.
Metformin: Souběžné podávání více dávek metforminu 1 000 mg se sitagliptinem 50 mg dvakrát denně významně nezměnilo farmakokinetiku sitagliptinu u pacientů s diabetem 2. typu.
Cyklosporin: Byla provedena studie hodnotící účinek cyklosporinu, silného inhibitoru p-glykoproteinu, na farmakokinetiku sitagliptinu.Současné podání jedné perorální dávky 100 mg sitagliptinu a jedné perorální dávky 600 mg cyklosporinu zvýšilo AUC a Cmax sitagliptinu přibližně o 29%, respektive 68%. Tyto změny ve farmakokinetice sitagliptinu nebyly považovány za klinicky relevantní. Renální clearance sitagliptinu nebyla významně změněna. Proto se neočekávají žádné interakce, relevantní s jinými inhibitory p-glykoproteinu.
Účinky sitagliptinu na jiné léčivé přípravky
Digoxin: Sitagliptin měl omezený účinek na plazmatické koncentrace digoxinu. Po podání 0,25 mg digoxinu souběžně se 100 mg sitagliptinu denně po dobu 10 dnů se plazmatická AUC digoxinu zvýšila v průměru o 11%a plazmatická Cmax v průměru o 18%. Nedoporučuje se úprava dávky digoxinu. Při současném podávání sitagliptinu a digoxinu by však měla být monitorována toxicita digoxinu u pacientů s rizikem toxicity digoxinu.
Data in vitro naznačují, že sitagliptin neinhibuje ani neindukuje izoenzymy CYP450. V klinických studiích sitagliptin významně nezměnil farmakokinetiku metforminu, glyburidu, simvastatinu, rosiglitazonu, warfarinu nebo perorálních kontraceptiv, což poskytuje důkaz in vivonízká náchylnost k interakcím se substráty CYP3A4, CYP2C8, CYP2C9 a s transportérem organických kationtů (OCT). Sitagliptin může být slabým inhibitorem p-glykoproteinu in vivo.
04.6 Těhotenství a kojení
Těhotenství
Adekvátní údaje o podávání sitagliptinu těhotným ženám nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu při vysokých dávkách (viz bod 5.3). Potenciální riziko pro člověka není známo. Vzhledem k nedostatku údajů u lidí by přípravek Januvia neměl být používán během těhotenství.
Čas krmení
Není známo, zda se sitagliptin vylučuje do lidského mléka. Studie na zvířatech prokázaly vylučování sitagliptinu do mateřského mléka. Januvia by neměla být používána během kojení.
Plodnost
Údaje získané na zvířatech nenaznačují účinek léčby sitagliptinem na mužskou a ženskou plodnost. Údaje o lidech chybí.
04.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Januvia nemá žádný nebo má zanedbatelný vliv na schopnost řídit a obsluhovat stroje. Při řízení vozidel nebo obsluze strojů je však třeba mít na paměti, že byly hlášeny závratě a ospalost.
Kromě toho, pokud je přípravek Januvia používán v kombinaci se sulfonylureou nebo s inzulínem, pacienti by měli být upozorněni na riziko hypoglykémie.
04.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Byly hlášeny závažné nežádoucí účinky včetně pankreatitidy a reakcí z přecitlivělosti.
V souvislosti se sulfonylureou (4,7%-13,8%) a inzulinem (9,6%) byla hlášena hypoglykémie (viz bod 4.4).
Tabulka nežádoucích účinků
Nežádoucí účinky jsou uvedeny níže (tabulka 1) podle třídy orgánových systémů a frekvence. Četnosti jsou definovány jako: velmi časté (≥ 1/10); časté (≥ 1/100,
Tabulka 1. Frekvence nežádoucích účinků zjištěných z placebem kontrolovaných klinických studií s monoterapií sitagliptinem a ze zkušeností po uvedení přípravku na trh
* Nežádoucí účinky, které byly zjištěny při postmarketingovém sledování.
† Viz bod 4.4.
Popis vybraných nežádoucích účinků
Kromě výše popsaných nežádoucích účinků souvisejících s léčivem zahrnovaly nežádoucí účinky hlášené bez ohledu na příčinnou souvislost s léčivým přípravkem, které se vyskytly nejméně v 5% případů a nejčastěji u pacientů léčených sitagliptinem, infekci horních cest dýchacích a nazofaryngitidu. Další nežádoucí účinky hlášené bez ohledu na příčinnou souvislost s léčivým přípravkem, které se vyskytovaly častěji u pacientů léčených sitagliptinem (které nedosahovaly 5% hladiny, ale vyskytovaly se s incidencí> 0,5% vyšší u sitagliptinu oproti kontrolní skupině ) zahrnovaly osteoartrózu a bolest v končetinách.
Některé nežádoucí účinky byly pozorovány častěji ve studiích kombinované léčby sitagliptinem s jinými antidiabetiky, než ve studiích monoterapie sitagliptinem. Jednalo se o hypoglykémii (velmi časté u kombinace sulfonylmočoviny a metforminu), chřipku (časté u inzulinu (s nebo bez) metformin), nauzea a zvracení (časté u metforminu), flatulence (časté u metforminu nebo pioglitazonu), zácpa (časté u kombinace sulfonylmočoviny a metforminu), periferní edém (časté u pioglitazonu nebo u kombinace pioglitazonu a metforminu) somnolence a průjem (méně časté u metforminu) a sucho v ústech (méně časté u inzulinu (s nebo bez metforminu)).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky, které se vyskytnou po registraci léčivého přípravku, je důležité, protože umožňuje průběžné sledování poměru přínosů a rizik léčivého přípravku. Zdravotničtí pracovníci jsou požádáni, aby hlásili jakékoli podezření na nežádoucí účinky prostřednictvím italské agentury pro léčivé přípravky. , webové stránky: www.agenziafarmaco.gov.it/it/responsabili.
04.9 Předávkování
Během kontrolovaných klinických studií na zdravých subjektech byly podány jednotlivé dávky sitagliptinu až do 800 mg. Minimální zvýšení QTc, které nebylo považováno za klinicky relevantní, bylo pozorováno při dávce sitagliptinu 800 mg v jedné studii. V klinických studiích nejsou žádné zkušenosti s dávkami vyššími než 800 mg. Ve studiích fáze I s opakovanými dávkami s dávkami sitagliptinu až 600 mg denně po dobu až 10 dnů a 400 mg denně po dobu až 28 dní nebyly pozorovány žádné dávkově závislé reakce.
V případě předávkování je rozumné použít běžná podpůrná opatření, např .: odstranit neabsorbovaný materiál z gastrointestinálního traktu, použít klinické sledování (včetně elektrokardiografie) a v případě potřeby zahájit podpůrnou péči.
Dialyzovatelnost sitagliptinu je mírná. V klinických studiích bylo během 3-4hodinové hemodialýzy odebráno přibližně 13,5% dávky. Pokud je to klinicky vhodné, lze zvážit prodlouženou hemodialýzu. Dialyzovatelnost sitagliptinu s peritoneální dialýzou není známa.
05.0 FARMAKOLOGICKÉ VLASTNOSTI
05.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: léky používané při diabetu, inhibitory dipeptidyl peptidázy 4 (DPP-4).
ATC kód: A10BH01.
Mechanismus účinku
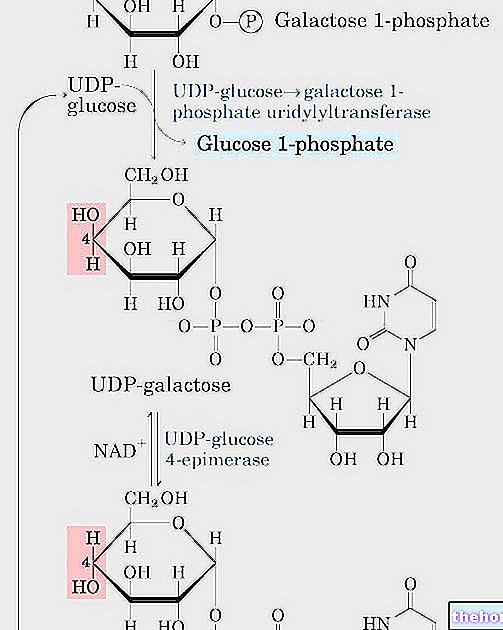
Januvia patří do třídy perorálních antihyperglykemických léků nazývaných inhibitory dipeptidyl peptidázy 4 (DPP-4). Zlepšení kontroly glykémie pozorované u tohoto léčivého přípravku může být zprostředkováno „zvýšenými hladinami aktivních inkretinů. Inkretiny, hormony, které zahrnují glukagonu podobný peptid-1 (GLP-1) a na glukóze závislý inzulinotropní polypeptid (GIP)“, jsou uvolňují se ze střeva během dne a jejich hladina se zvyšuje v reakci na jídlo. Inkretiny jsou součástí endogenního systému zapojeného do fyziologické regulace homeostázy glukózy. Pokud je hladina glukózy v krvi normální nebo zvýšená, GLP-1 a GIP zvyšují syntézu a uvolňování inzulínu pankreatickými beta buňkami intracelulárními signálními cestami zahrnujícími cyklický AMP.Léčba inhibitory GLP-1 nebo DPP-4 na zvířecích modelech diabetu 2. typu prokázala zlepšení reakce beta buněk na glukózu a stimulaci biosyntézy a uvolňování inzulínu. Při vyšších hladinách inzulínu se zvyšuje vychytávání tkáňové glukózy. GLP-1 také snižuje sekreci glukagonu pankreatickými alfa buňkami. Nižší koncentrace glukagonu spolu s vyššími hladinami inzulínu mají za následek sníženou produkci glukózy v játrech, což má za následek snížení hladiny glukózy v krvi. Účinky GLP-1 a GIP jsou závislé na glukóze, takže když je hladina glukózy v krvi nízká, nejsou pozorovány žádné podněty pro uvolňování inzulínu a potlačení sekrece glukagonu. U GLP-1 i GIP se stimulace uvolňování inzulinu zvyšuje, když hladina glukózy stoupne nad normální hodnotu Kromě toho GLP-1 neovlivňuje normální reakci glukagonu na hypoglykémii. Aktivita GLP-1 a GIP je omezena enzymem DPP-4, který rychle hydrolyzuje inkretiny na neaktivní metabolity. Sitagliptin zabraňuje hydrolýze inkretinů DPP-4, čímž zvyšuje plazmatické koncentrace aktivních forem GLP-1 a GIP. Zvýšení hladin aktivních inkretinů sitagliptin zvyšuje uvolňování inzulínu a snižuje hladinu glukagonu glukózovým způsobem. U pacientů s typem 2 diabetes s hyperglykemií, tyto změny v hladinách inzulínu a glukagonu vedou ke snížení hemoglobinu A1c (HbA1c) a nižším koncentracím glukózy nalačno a v krvi. post prandium. Mechanismus sitagliptinu závislý na glukóze je odlišný od mechanismu derivátů sulfonylmočoviny, které zvyšují sekreci inzulínu, i když jsou hladiny glukózy nízké, a mohou vést k hypoglykémii u pacientů s diabetem typu 2 a u normálních subjektů. Sitagliptin je silný a vysoce selektivní inhibitor Enzym DPP-4 a při terapeutických koncentracích neinhibuje aktivitu blízce příbuzných enzymů DPP-8 nebo DPP-9.
Ve 2denní studii na zdravých subjektech samotný sitagliptin zvyšoval koncentrace aktivního GLP-1, zatímco samotný metformin zvyšoval aktivní i celkový GLP-1 koncentrace podobně. Souběžné podávání sitagliptinu a metforminu mělo aditivní účinek na aktivní koncentrace GLP-1. Sitagliptin, ale ne metformin, zvýšil aktivní koncentrace GIP.
Klinická účinnost a bezpečnost
Celkově sitagliptin zlepšil kontrolu glykémie, pokud byl podáván v monoterapii nebo v kombinované terapii (viz tabulka 2).
Byly provedeny dvě studie k vyhodnocení účinnosti a bezpečnosti samotného sitagliptinu. Léčba monoterapií sitagliptinem v dávce 100 mg jednou denně vedla k významnému zlepšení HbA1c, plazmatické glukózy nalačno (FPG) a 2hodinové postprandiální glukózy (2hodinové PPG) ve srovnání s placebem ve dvou studiích, jedna trvala 18 týdnů a druhá 24 týdnů. Zlepšení náhradních markerů funkce beta buněk, včetně HOMA-β (Homeostasis Model Assessment-β), poměru proinzulinu / inzulínu a míry odpovědi beta buněk na test tolerance jídla při častém odběru vzorků. Pozorovaný výskyt hypoglykémie u pacientů léčených sitagliptinem byl podobný placebu. Tělesná hmotnost ve dvou studiích se při léčbě sitagliptinem oproti výchozím hodnotám nezvyšovala ve srovnání s mírným poklesem pozorovaným u pacientů léčených placebem.
Sitagliptin 100 mg jednou denně vyvolal významné zlepšení glykemických parametrů ve srovnání s placebem ve dvou 24týdenních doplňkových studiích sitagliptinu, jedna v kombinaci s metforminem a jedna v kombinaci s pioglitazonem. Změna tělesné hmotnosti od výchozí hodnoty byla podobná u pacientů léčených sitagliptinem ve srovnání s placebem. V těchto studiích byl „a“ podobný výskyt hypoglykémie hlášen u pacientů léčených sitagliptinem nebo placebem.
24týdenní placebem kontrolovaná studie byla navržena tak, aby vyhodnotila účinnost a bezpečnost sitagliptinu (100 mg jednou denně) přidaného k samotnému glimepiridu nebo ke glimepiridu v kombinaci s metforminem. Přidání sitagliptinu nebo glimepiridu samotného nebo s glimepiridem a metforminem vyvolalo významné zlepšení glykemických parametrů. Pacienti léčení sitagliptinem měli mírný nárůst tělesné hmotnosti ve srovnání s těmi, kterým bylo podáváno placebo.
26týdenní placebem kontrolovaná studie byla navržena tak, aby vyhodnotila účinnost a bezpečnost sitagliptinu (100 mg jednou denně) přidaného ke kombinaci pioglitazonu a metforminu. Přidání sitagliptinu k pioglitazonu a metforminu vedlo k významnému zlepšení glykemických parametrů. Změna tělesné hmotnosti oproti výchozím hodnotám byla podobná u pacientů léčených sitagliptinem a u pacientů léčených placebem. Výskyt hypoglykémie byl také podobný u pacientů léčených placebem pacienti léčení sitagliptinem nebo placebem.
24týdenní placebem kontrolovaná studie byla navržena tak, aby vyhodnotila účinnost a bezpečnost sitagliptinu (100 mg jednou denně) přidaného k inzulinu (ve stabilní dávce po dobu alespoň 10 týdnů) s nebo bez metforminu (alespoň 1 500 mg.). U pacientů užívajících premixovaný inzulín byla průměrná denní dávka 70,9 U / den. U pacientů užívajících nemixovaný (středně působící / dlouhodobě působící) inzulín byla průměrná denní dávka 44,3 U / den. Přidání sitagliptinu k inzulinu vyvolalo významné zlepšení glykemických parametrů. V žádné skupině nedošlo k žádné významné změně tělesné hmotnosti od výchozí hodnoty.
Ve 24týdenní, placebem kontrolované, z faktoriální studie zahajovací kombinační terapie, sitagliptin 50 mg dvakrát denně v kombinaci s metforminem (500 mg nebo 1 000 mg dvakrát denně) vedl k významnému zlepšení glykemických parametrů ve srovnání s každou monoterapií. s kombinací sitagliptin a metformin byl podobný tomu, který byl pozorován u samotného metforminu nebo u placeba; u pacientů léčených monoterapií sitagliptinem nebyla pozorována žádná změna oproti výchozímu stavu. Výskyt hypoglykemie byl mezi léčenými skupinami podobný.
Tabulka 2: Výsledky HbA1c ve studiích s placebem kontrolovanou monoterapií a kombinovanou terapií *
24týdenní aktivně kontrolovaná studie (metformin) byla navržena tak, aby vyhodnotila účinnost a bezpečnost sitagliptinu 100 mg jednou denně (N = 528) ve srovnání s metforminem (N = 522) u pacientů, kteří neměli dostatečnou glykemickou kontrolu dietou a cvičení a kteří nebyli na antihyperglykemické terapii (bez terapie po dobu alespoň 4 měsíců). Průměrná dávka metforminu byla přibližně 1 900 mg denně. Snížení HbA1c z průměrných výchozích hodnot 7,2% bylo -0,43% pro sitagliptin a -0,57% pro metformin (analýza podle protokolu). Celková incidence gastrointestinálních nežádoucích účinků považovaných za související s léčivem u pacientů léčených sitagliptinem byla 2,7% ve srovnání s 12,6% u pacientů léčených metforminem. Výskyt hypoglykémie se významně nelišil mezi léčenými skupinami (sitagliptin, 1,3%; metformin, 1,9%). V obou skupinách se tělesná hmotnost snížila od výchozí hodnoty (sitagliptin, -0,6 kg; metformin -1,9 kg).
Ve studii porovnávající účinnost a bezpečnost přidání 100 mg sitagliptinu jednou denně nebo glipizidu (sulfonylmočoviny) u pacientů s nedostatečnou kontrolou glykémie při monoterapii metforminem byl sitagliptin při snižování HbA1c podobný glipizidu. Průměrná dávka glipizidu použitá ve srovnávací skupině byla 10 mg / den, přičemž přibližně 40% pacientů vyžadovalo po celou dobu studie dávku glipizidu ≤ 5 mg / den. Pacienti ve skupině se sitagliptinem však zaznamenali více přerušení z důvodu nedostatečné účinnosti než ve skupině s glipizidem. Pacienti léčení sitagliptinem vykazovali významný průměrný pokles tělesné hmotnosti oproti výchozím hodnotám ve srovnání s významným nárůstem hmotnosti pozorovaným u pacientů užívajících glipizid (-1,5 vs + 1,1 kg). V této studii se poměr proinzulinu / inzulinu, ukazatele syntézy inzulínu a účinnosti uvolňování, zlepšil se sitagliptinem a zhoršil se léčbou glipizidem. Výskyt hypoglykemie ve skupině se sitagliptinem (4,9%) byl významně nižší než ve skupině s glipizidem (32,0%).
24týdenní placebem kontrolovaná studie zahrnující 660 pacientů byla navržena tak, aby vyhodnotila účinnost a bezpečnost šetřící inzulín sitagliptinu (100 mg jednou denně) přidaného k inzulinu glargin s nebo bez metforminu (nejméně 1 500 mg) během intenzifikace inzulínové terapie. Výchozí hodnota HbA1c byla 8,74% a výchozí dávka inzulínu byla 37 IU / den. Pacienti byli instruováni, aby titrovali dávku inzulínu glargin na základě hodnot glukózy nalačno měřených prstem. Ve 24. týdnu bylo zvýšení denní dávky inzulinu o 19 IU / den u pacientů léčených sitagliptinem a 24 IU / den u pacientů léčených placebem. Snížení HbA1c u pacientů léčených sitagliptinem a inzulínem (s nebo bez metforminu) bylo - 1,31% oproti -0,87% u pacientů léčených placebem a inzulínem (s nebo bez metforminu), rozdíl -0,45% [95% CI: -0,60, -0,29]. Výskyt hypoglykemie byl 25,2% u pacientů léčených sitagliptinem a inzulín (s nebo bez metforminu) a 36,8% u pacientů léčených placebem a inzulínem (s nebo bez metforminu). Rozdíl byl způsoben především vyšším procentem pacientů ve skupině s placebem, u kterých se vyskytly 3 nebo více epizod hypoglykémie (9,4 vs. 19,2%). Nebyl žádný rozdíl ve výskytu těžké hypoglykemie.
U pacientů se středně těžkou až těžkou poruchou funkce ledvin byla provedena studie porovnávající sitagliptin 25 nebo 50 mg jednou denně a glipizid 2,5 až 20 mg / den. Tato studie zahrnovala 423 pacientů s chronickým poškozením ledvin (odhadovaná rychlost glomerulární filtrace
Další studie porovnávající sitagliptin 25 mg jednou denně a glipizid 2,5 až 20 mg / den byla provedena u 129 pacientů s ESRD, kteří byli na dialýze. Po 54 týdnech bylo průměrné snížení HbA1c oproti výchozím hodnotám -0,72% u sitagliptinu a -0,87% u glipizidu. V této studii byl profil účinnosti a bezpečnosti sitagliptinu 25 mg jednou denně obecně podobný profilu pozorovanému v jiných studiích monoterapie prováděných u pacientů s normální funkcí ledvin. Výskyt hypoglykémie nebyl významně odlišný mezi léčenými skupinami (sitagliptin, 6,3%; glipizid, 10,8%).
V jiné studii zahrnující 91 pacientů s diabetem 2. typu a chronickým poškozením ledvin (clearance kreatininu
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Januvia u jedné nebo více podskupin pediatrické populace s diabetes mellitus 2. typu (informace o použití u dětí viz bod 4.2).
05,2 "Farmakokinetické vlastnosti
Vstřebávání
Po perorálním podání dávky 100 mg zdravým subjektům byl sitagliptin rychle absorbován, s maximálními plazmatickými koncentracemi (medián Tmax) 1 až 4 hodiny po podání dávky, průměrná plazmatická AUC sitagliptinu byla 8,52 mcM • nyní byla Cmax 950 nM. Absolutní biologická dostupnost sitagliptinu je přibližně 87%. Protože souběžné podávání jídla s vysokým obsahem tuku se sitagliptinem nemělo žádný vliv na farmakokinetiku, lze přípravek Januvia podávat nezávisle na jídle.
Plazmatická AUC sitagliptinu se zvyšovala způsobem úměrným dávce. Úměrnost dávky nebyla stanovena pro Cmax a C24h (Cmax se zvýšila více než úměrnost dávce a C24h se zvýšila v menší míře. S ohledem na úměrnost dávce).
Rozdělení
Průměrný distribuční objem v ustáleném stavu po jednorázové intravenózní dávce 100 mg sitagliptinu zdravým subjektům je přibližně 198 litrů. Podíl sitagliptinu navázaného na plazmatické proteiny reverzibilním způsobem je nízký (38%).
Biotransformace
Sitagliptin je eliminován beze změny primárně močí a metabolismus je malou metabolickou cestou. Přibližně 79% sitagliptinu se vylučuje v nezměněné formě močí.
Po perorální dávce [14C] sitagliptinu bylo přibližně 16% radioaktivity vyloučeno jako metabolity sitagliptinu. Byly nalezeny stopy šesti metabolitů sitagliptinu a neočekává se, že by přispívaly k plazmatické inhibiční aktivitě sitagliptinu vůči DPP-4. in vitro ukázal, že enzym primárně zodpovědný za omezený metabolismus sitagliptinu je CYP3A4, s přispěním CYP2C8.
Data in vitro ukázal, že sitagliptin není inhibitorem izoenzymů CYP CYP3A4, 2C8, 2C9, 2D6, 1A2, 2C19 nebo 2B6 a není induktorem CYP3A4 a CYP1A2.
Odstranění
Po orálním podání [14C] sitagliptinu zdravým subjektům bylo přibližně 100%podané radioaktivity vyloučeno stolicí (13%) nebo močí (87%) do jednoho týdne po podání. Zdánlivý terminální t½ po 100 mg perorální dávce sitagliptinu byl přibližně 12,4 hodiny. Sitagliptin se při více dávkách hromadí jen minimálně. Renální clearance byla přibližně 350 ml / min.
Eliminace sitagliptinu probíhá především renální exkrecí a zahrnuje aktivní tubulární sekreci. Sitagliptin je substrátem transportéru lidského organického aniontu 3 (hOAT-3), který se může podílet na renální eliminaci sitagliptinu. Klinický význam hOAT-3 pro transport sitagliptinu nebyl stanoven. Sitagliptin je také substrátem p -glykoprotein, který se také může podílet na zprostředkování renální eliminace sitagliptinu. Cyklosporin, inhibitor p-glykoproteinu, však nesnížil renální clearance sitagliptinu. Sitagliptin není substrátem pro transportéry OCT2 nebo OAT1 nebo PEPT½. In vitrositagliptin neinhiboval transport zprostředkovaný OAT3 (IC50 = 160 mcM) nebo p-glykoproteinem (až 250 mcM) při terapeuticky relevantních plazmatických koncentracích. V klinické studii měl sitagliptin omezený účinek na plazmatické koncentrace digoxinu, což naznačuje, že sitagliptin může být slabým inhibitorem p-glykoproteinu.
Charakteristika pacientů
Farmakokinetika sitagliptinu byla u zdravých subjektů a pacientů s diabetem 2. typu obecně podobná.
Poškození ledvin
Byla provedena otevřená studie s jednorázovou dávkou k vyhodnocení farmakokinetiky snížené dávky sitagliptinu (50 mg) u pacientů s různým stupněm chronického poškození ledvin ve srovnání s normálními zdravými kontrolními subjekty. Studie zahrnovala pacienty s poruchou funkce ledvin klasifikovanou podle clearance kreatininu jako mírnou (50 až
U pacientů s mírnou poruchou funkce ledvin nedošlo ke klinicky významnému zvýšení plazmatických koncentrací sitagliptinu ve srovnání s normálními zdravými kontrolními subjekty. U pacientů se středně těžkou poruchou funkce ledvin bylo pozorováno přibližně 2násobné zvýšení plazmatické AUC sitagliptinu a u pacientů s těžkou poruchou funkce ledvin a ESDR na hemodialýze bylo pozorováno přibližně 4násobné zvýšení plazmatické AUC ve srovnání se zdravými kontrolními subjekty. Sitagliptin byl v omezené míře odstraněn hemodialýzou (13,5% během 3 až 4 hodinové hemodialýzy se zahájením 4 hodiny po podání dávky). Aby se dosáhlo plazmatických koncentrací sitagliptinu podobných koncentracím zjištěným u pacientů s normální funkcí ledvin, doporučují se nižší dávky pacienti se středně těžkou a těžkou poruchou funkce ledvin, stejně jako u pacientů s ESRD vyžadujícím dialýzu (viz bod 4.2).
Porucha funkce jater
U pacientů s lehkou nebo středně těžkou poruchou funkce jater (Child-Pugh skóre ≤ 9) není pro přípravek Januvia nutná žádná úprava dávky. U pacientů s těžkou poruchou funkce jater (Child-Pughovo skóre> 9) nejsou žádné klinické zkušenosti. Protože je však sitagliptin primárně vylučován ledvinami, neočekává se, že by závažné poškození jater ovlivnilo farmakokinetiku sitagliptinu.
Senioři
Na základě věku není nutná úprava dávky. Věk neměl žádný klinicky významný dopad na farmakokinetiku sitagliptinu na základě údajů z farmakokinetické analýzy populace fáze I a fáze II. U starších osob (od 65 do 80 let) přibližně o 19% vyšší plazma byly pozorovány koncentrace sitagliptinu než u mladých lidí.
Pediatrická populace
Nebyly provedeny žádné studie s přípravkem Januvia u pediatrických pacientů.
Další charakteristiky pacientů
Na základě pohlaví, rasy nebo indexu tělesné hmotnosti (BMI) není nutná úprava dávky. Tyto charakteristiky neměly klinicky významný účinek na farmakokinetiku sitagliptinu na základě údajů z kompozitní farmakokinetické analýzy fáze I a údajů z populační farmakokinetické analýzy fáze I a fáze II.
05.3 Předklinické údaje vztahující se k bezpečnosti
U hlodavců byla renální a jaterní toxicita pozorována při systémových expozičních hodnotách rovnajících se 58násobku lidské expozice, zatímco při 19násobku lidské expozice nebyla zjištěna úroveň žádného účinku. Abnormality řezáku byly pozorovány u potkanů při úrovních expozice rovnajících se 67násobku klinické expozice člověka; úroveň bez účinku pro tuto událost byla 58krát vyšší na základě 14týdenní studie na potkanech. Relevance těchto údajů pro člověka není známa. U psů byly pozorovány přechodné fyzické příznaky související s léčbou při expozičních hladinách přibližně 23krát vyšších než je klinická expoziční úroveň, z nichž některé naznačují nervovou toxicitu, jako je dýchání otevřenými ústy, slinění, bílá pěnivost zvracení, ataxie, třes, snížená aktivita a / nebo ohnuté držení těla. Při dávkách odpovídajících přibližně 23násobku úrovně systémové expozice u lidí byla histologicky pozorována také velmi mírná až mírná degenerace kosterního svalstva. Při expozici rovnající se 6násobku úrovně klinické expozice nebyla zjištěna úroveň účinku pro tyto příhody.
Sitagliptin v předklinických studiích neprokázal genotoxicitu. Sitagliptin nebyl u myší karcinogenní. U potkanů došlo ke zvýšení výskytu jaterních adenomů a karcinomů při systémových expozičních hladinách rovných 58násobku expozice člověka. Jelikož bylo prokázáno, že hepatotoxicita koreluje s indukcí rakoviny jater u potkanů, tento zvýšený výskyt nádorů jater u krysa je pravděpodobně sekundární k chronické jaterní toxicitě vyskytující se při těchto vysokých dávkách.
Vzhledem k velkému rozpětí bezpečnosti (19krát na této úrovni bez účinku) nejsou tyto neoplastické léze považovány za relevantní vzhledem k expozičním okolnostem u lidí.
U samců a samic potkanů léčených sitagliptinem před a během páření nebyly pozorovány žádné nežádoucí účinky na plodnost.
Ve studiích prenatálního / postnatálního vývoje provedených na potkanech sitagliptin neprokázal žádné nežádoucí účinky.
Studie reprodukční toxicity prokázaly mírné zvýšení výskytu malformací žeber plodu (chybějící, hypoplastická a vlnitá žebra) související s léčbou u potomků potkanů při systémových expozičních hladinách 29krát vyšších než jsou úrovně expozice člověka. Mateřská toxicita byla pozorována u králíků při expozičních hladinách větších než 29násobek expozičních úrovní u lidí.Vzhledem k širokému bezpečnostnímu rozpětí tato zjištění nenaznačují přítomnost příslušných reprodukčních rizik u lidí. Sitagliptin se vylučuje ve značných množstvích do mléka kojících potkanů (poměr mléko / plazma: 4: 1).
06.0 FARMACEUTICKÉ INFORMACE
06.1 Pomocné látky
Jádro tabletu:
mikrokrystalická celulóza (E460);
bezvodý hydrogenfosforečnan vápenatý (E341);
sodná sůl kroskarmelózy (E468);
stearát hořečnatý (E470b);
natrium -stearyl -fumarát.
Potah tablety:
poly (vinylalkohol);
makrogol 3350;
mastek (E553b);
oxid titaničitý (E171);
červený oxid železitý (E172);
žlutý oxid železitý (E172).
06.2 Neslučitelnost
Irelevantní.
06.3 Doba platnosti
3 roky.
06.4 Zvláštní opatření pro skladování
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
06.5 Charakter vnitřního obalu a obsah balení
Neprůhledné blistry (PVC / PE / PVDC a hliník). Balení 14, 28, 30, 56, 84, 90 nebo 98 potahovaných tablet a 50 x 1 potahovaných tablet v perforovaných jednodávkových blistrech.
Na trhu nemusí být všechny velikosti balení.
06.6 Návod k použití a zacházení
Nepoužitý léčivý přípravek a odpad z tohoto přípravku musí být zlikvidován v souladu s místními předpisy.
07.0 DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon
Hertfordshire EN11 9BU
Spojené království
08.0 REGISTRAČNÍ ČÍSLO
EU/1/07/383/001
037793015
EU/1/07/383/002
037793027
EU/1/07/383/003
037793039
EU/1/07/383/004
037793041
EU/1/07/383/005
037793054
EU/1/07/383/006
037793066
EU/1/07/383/019
037793193
EU/1/07/383/020
037793205
09.0 DATUM PRVNÍ REGISTRACE NEBO PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. března 2007
Datum posledního obnovení: 21. března 2012
10.0 DATUM REVIZE TEXTU
28. května 2015